Hintergrund
Das Alice im Wunderland-Syndrom (AIWS) ist eine seltene neurologische Störung, die durch Verzerrungen der visuellen Wahrnehmung (Metamorphopsien), des Körperbildes und der Erfahrung der Zeit gekennzeichnet ist. Wie bereits 1955 von John Todd festgestellt, können diese Symptome von Derealisierung und Depersonalisierung begleitet sein (1)., Patienten, die an AIWS leiden, können am Ende einen Neurologen oder Psychiater konsultieren, obwohl es in beiden Fachgebieten nicht so bekannt ist, wie es es verdient. Dies ist zumindest teilweise darauf zurückzuführen, dass wichtige Klassifikationen wie das diagnostische und statistische Handbuch für psychische Störungen es nicht als diagnostische Kategorie auflisten, während andere, wie die Internationale Klassifikation von Krankheiten, ihr nur begrenzte Aufmerksamkeit schenken. Ein weiterer Grund kann die relativ geringe Anzahl veröffentlichter Fälle sein., Eine Überprüfung der vorhandenen Literatur, die 2016 veröffentlicht wurde, ergab, dass seit der Konzeptualisierung des Syndroms im Jahr 1955 nur 169 Fälle von AIWS beschrieben wurden, was auf eine mittlere Anzahl von 1, 1 Fällen pro Jahr hinausläuft (4). Obwohl diese Zahl in den letzten Jahren stetig gestiegen ist, liegt sie derzeit bei rund 180. Dies sind alles Fälle, die ärztliche Hilfe benötigen, d. H. Was wir „klinische Fälle“ nennen.“In der Zwischenzeit zeigen groß angelegte Bevölkerungsstudien, dass Symptome von AIWS in der Allgemeinbevölkerung häufig auftreten, mit lebenslangen Prävalenzraten von bis zu 30% (5-7)., Da letztere Fälle typischerweise flüchtig und vorübergehend sind und selten hilfesuchendes Verhalten mit sich bringen, werden sie als „nichtklinische Fälle“ bezeichnet.“Von den klinischen Fällen zeigen etwa 85% nur eine einzige sensorische Modalität. Von ihnen sind die meisten auf ein einziges Symptom beschränkt (z. B. nur Mikropsie oder nur Makrosomatognosie usw.).) (8). AIWS hat viele verschiedene Ursachen. In klinischen Fällen ist Enzephalitis (insbesondere aufgrund einer Epstein-Barr-Virusinfektion) die häufigste Ursache bei Kindern; bei Erwachsenen ist es Migräne (4)., Obwohl Prognose und Ergebnis weitgehend von der zugrunde liegenden Erkrankung und ihrer Behandlbarkeit abhängen, kann die durch AIWS verursachte Belastung erheblich sein, selbst wenn ihre Ursache ziemlich harmlos ist, wie im Zusammenhang mit Fieber oder sporadischem Cannabiskonsum. Dies gilt insbesondere dann, wenn Symptome die Sinneswahrnehmung stark stören, die tägliche Funktion beeinträchtigen und/oder katastrophale Gedanken hervorrufen (z. B. dass sich die Welt tatsächlich unverständlich verändert hat oder dass Symptome auf Demenz oder Schizophrenie hindeuten können)., Da nichtklinische Fälle per Definition selbstlimitierend sind und selbst in 50% aller klinischen Fälle eine Beruhigung zu genügen scheint, wird AIWS manchmal als relativ harmlos bezeichnet (9). Da die Forschung auf diesem Gebiet jedoch noch in den Kinderschuhen steckt und AIWS möglicherweise stark unterreportiert werden, ist es zu früh, um Rückschlüsse auf ihre Naturgeschichte und ihr „typisches“ Ergebnis zu ziehen. Darüber hinaus wissen wir bereits, dass AIWS eine Manifestation schwerer und sogar tödlicher Zustände wie Hirninfarkt oder Hirntumor sein kann (10, 11)., Wir stellen hier einen Fall von AIWS vor, der durch die sporadische Creutzfeldt-Jakob-Krankheit (CJD) verursacht wird. Damit fügen wir der aufkeimenden Literatur über AIWS hinzu und versuchen, vorzeitigen Schlussfolgerungen entgegenzuwirken, dass dieses Syndrom a priori als relativ „gutartig“ charakterisiert werden kann.
Methoden
Der von uns beschriebene Patient wurde in unserer eigenen klinischen Praxis behandelt. Da er innerhalb von 2 Monaten nach Beginn seiner Symptome von AIWS starb, erhielten wir von seiner Familie die schriftliche Zustimmung zur Veröffentlichung.,
Ergebnisse-Fallbericht
Im März 2018 meldete sich ein 68-jähriger Mann mit Sehsymptomen in unserer neurologischen Ambulanz. Er hatte eine Geschichte von Kataraktchirurgie im Jahr 2004 und Wahrnehmung Taubheit ab 2006. Ein paar Wochen bevor wir ihn sahen, war er wegen schlechter Tiefenund Größenwahrnehmung und eines angespannten Gefühls um den Kopf, das 1 Monat lang bestanden hatte, an den Augenarzt überwiesen worden. Er hatte keine anderen (neurologischen) Symptome. Die ophthalmologische Untersuchung ergab ein Offenwinkelglaukom mit Augeninnendruck von 39/27 mmHg ODS (Referenz 12-21 mmHg)., Seine Sehschärfe war nahezu normal (OD 0·8 dpt, OS 0·9 dpt), obwohl die Gesichtsfelduntersuchung mehrere leichte Defekte (OD) zeigte, die nicht mit seinen Symptomen übereinstimmten. Sein Augendruck normalisierte sich mit der Verwendung von Bimatoprost und Dorzolamid/Timolol Augentropfen. Im Laufe der folgenden Wochen wurden die visuellen Symptome jedoch ausgeprägter und eigenartiger. Bei der Überweisung an unsere Ambulanz berichtete er, dass sich bewegende Objekte verlangsamten und beschleunigten „wie in einem Charlie Chaplin-Film“ (bekannt als langwierige Dauer bzw.,, varianten der Gruppe der Zeitverzerrungen). Manchmal sah er überhaupt keine Bewegung (Akinetopsie). Darüber hinaus empfand er Farben als außergewöhnlich hell (Hyperchromatopsie) und wechselte zufällig, wie ein rotes T-Shirt, das grün wurde „wie Fairground Lights“ (Chloropsie/Dyschromatopsie). Er sah auch Objekte schrumpfen oder auf eine unnatürliche Größe anschwellen, „als würde er in einen Funhouse-Spiegel schauen“ (Mikropsie und Makropsie, Zoom Vision)., Zusätzlich zu diesen Arten von Metamorphopsie entwickelte er Parästhesien in Händen und Füßen, Ganginstabilität sowie Konzentrations-und Gedächtnisstörungen. Er hatte auch Schwierigkeiten, die Jazzflöte und das Saxophon zu spielen, was wir als bedrohliches Zeichen betrachteten, da er ein professioneller Jazzmusiker war. Seine Rede wurde weniger fließend, weil er nicht die richtigen Worte finden konnte oder sie nicht zu Sätzen zusammenfädelte, und sein Partner bemerkte Verhaltensänderungen in Form von Erregung und emotionaler Labilität.,
Obwohl neurologische Untersuchungen, routinemäßige Blutuntersuchungen und eine Kopf-MRT (einschließlich diffusionsgewichteter Bildgebung, DWI) keine Auffälligkeiten zeigten, wurde unser Patient zunehmend vergesslich und desorientiert und wurde anschließend in unser Krankenhaus eingeliefert. Dort sahen wir einen zuvor beredten Mann, der jetzt an Aphasie, Ataxie, myoklonischen Idioten und einer schnell fortschreitenden Demenz litt., Eine zweite Kopf-MRT zeigte einen niedrigen scheinbaren Diffusionskoeffizienten (ADC) und eine erhöhte DWI-Signalisierung im linken parieto-okzipitalen Kortex, charakteristisch für ein kortikales Bandzeichen (Abbildung 1), während das Elektroenzephalogramm periodische Scharfwellenkomplexe (PSWC) über derselben Fläche zeigte (Abbildung 2). Die MRT mit Vorhandensein von 14-3-3 Proteinen und erhöhtem Tau-Protein in der Zerebrospinalflüssigkeit unterstützte die Diagnose der wahrscheinlichen sporadischen Creutzfeldt-Jakob-Krankheit (CJD), insbesondere der Heidenhain-Variante., Wir haben andere mögliche Ursachen ausgeschlossen, einschließlich Autoimmunerkrankungen, Infektionen, Malignome, toxische und metabolische Enzephalopathien und andere neurodegenerative Erkrankungen (insbesondere posteriore kortikale Atrophie und Demenz mit Lewy-Körpern). Wir haben keine Gentests durchgeführt, da die Familienanamnese negativ für neurodegenerative Erkrankungen war, insbesondere CJD. Leider gab es keine therapeutischen Möglichkeiten. In Übereinstimmung mit seinen Wünschen wurde unser Patient nach Hause entlassen, wo er innerhalb von 2 Monaten nach Beginn seiner visuellen Symptome starb. Wir konnten keine Zustimmung zur Autopsie einholen.,
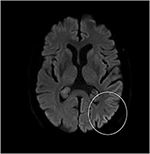
Abbildung 1. Hohe Signalintensität des linken parieto-okzipitalen Kortex bei diffusionsgewichteter MRT (DWI; b = 1.000).
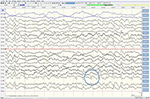
Abbildung 2. EEG zeigt periodische Scharfwellenkomplexe über der linken parieto-okzipitalen Region, die auf CJD hindeuten.,
Was diesen Fall einzigartig macht, ist, dass unser Patient eine relativ große Anzahl von Metamorphopsien hatte (was in der vorhandenen Literatur nur in 15% aller klinischen Fälle von AIWS berichtet wird) und dass CJD die wahrscheinlichste Ursache war. Da eine Diagnose einer bestimmten CJD nur post mortem durch Autopsie gestellt werden kann, war es bedauerlich, dass wir keine Zustimmung zur Autopsie erhalten konnten. Dennoch wurde festgestellt, dass die MRT-Ergebnisse in 83% der Fälle wahrscheinlicher CJD positiv sind (gegenüber 98% der Fälle bestimmter CJD) (12)., Obwohl die MRT des Kopfes nicht der Goldstandard ist, war eine Diagnose von CJD bei unserem Patienten immer noch sehr wahrscheinlich, insbesondere in Anbetracht unserer zusätzlichen neurologischen Aufarbeitung, die keine alternative Diagnose ergab, die das klinische Bild erklären könnte, einschließlich seines schnellen Rückgangs und dramatischen Ergebnisses.,
Alice im Wunderland-Syndrom
Wie John Todd 1955 beschrieb, ist AIWS durch Wahrnehmungsverzerrungen gekennzeichnet, die an die visuellen Verzerrungen, Zeitverzerrungen, körperlichen Veränderungen, Derealisierung und Depersonalisierung erinnern, die Alice während ihres Aufenthalts im Wunderland erlebt hat, wie in Lewis Carrolls klassischem Kinderbuch Alice ‚ s Adventures in Wonderland (1, 13) beschrieben., Todd, und vor ihm, Lipmann (14), schlug vor, dass Carroll–dessen richtiger Name Charles Lutwidge Dodgson war–Inspiration für diese besonderen Phänomene in Wahrnehmungsverzerrungen gefunden hatte, die er selbst im Zusammenhang mit Migräne erlebt hatte. Obwohl dieser Hypothese mehrere historische Abhandlungen folgten, in denen behauptet wurde, Migräne sei tatsächlich die wahrscheinlichste Quelle für Dodgsons Erfahrungen (15, 16), haben andere die ebenso verlockende Hypothese aufgestellt, dass es in seinem Fall eher Epilepsie war, die sie verursachte (17) oder vielleicht Drogenmissbrauch (18)., Obwohl wir wahrscheinlich nie sicher wissen werden, was Dodgson dazu gebracht hat, diese Symptome zu erleben, deutet eine sorgfältige Rekonstruktion seiner Krankengeschichte darauf hin, dass ihre wahrscheinlichste Quelle eine Infektionskrankheit war, an der der Autor von Kindheit an mehrfach litt, buchstäblich bis zum letzten Tag seines Lebens (19)., Es ist sehr wahrscheinlich, dass Dodgson diese Verzerrungen selbst erlebt hatte (anstatt die Erfahrungen seiner Heldin auf Informationen aus dritter Hand gestützt zu haben), mit satten 13 verschiedenen Symptomen, die im gesamten Buch beschrieben wurden, lange bevor Todd das Konzept von AIWS entwickelt hatte.,
Um zu verstehen, was AIWS von anderen Wahrnehmungssyndromen und-störungen abhebt, müssen wir erkennen, dass Verzerrungen sich von Halluzinationen und Illusionen dadurch unterscheiden, dass sie weder neu gebildete Perzepte von etwas sind, das nicht da ist (Halluzination), noch tatsächliche Objekte oder Szenen, die fälschlicherweise als etwas anderes beurteilt werden (Illusion). Stattdessen sind Verzerrungen Perzepte, die ein wachendes Individuum erlebt, die auf entsprechenden Reizen der Außenwelt beruhen, von denen ein hochspezifischer Aspekt konsistent verändert wird ., Heute sind etwa 55 verschiedene Arten von Wahrnehmungsverzerrungen bekannt, die im Zusammenhang mit AIWS auftreten können (4). Die am häufigsten berichteten sind Mikropsie (Dinge kleiner sehen als sie sind), Makropsie (Dinge größer sehen als sie sind), Teleopsie (Dinge weiter weg sehen als sie sind) und Dysmorphopsie (gerade Linien als wellig sehen). Die Bestimmung, dass solche Aspekte konsistent verändert werden, bedeutet, dass die Verzerrung für alles innerhalb des Sichtbereichs des Patienten gilt., Daher werden Menschen, die an Plagiopsie leiden, typischerweise alle Dinge als schräg, in die gleiche Richtung und in den gleichen Grad sehen, während diejenigen mit Prosopometamorphopsie typischerweise eine bestimmte Veränderung in allen Gesichtern sehen, die sie wahrnehmen, wie schwere Augenbrauen oder ein Auge, das deutlich an die Nase entführt wird, oder menschliche Gesichter, die sich in Drachengesichter verwandeln (21, 22). Die Art dieser Verzerrungen kann im Laufe der Zeit variieren, aber sobald sie vorhanden sind, erscheinen alle wahrgenommenen Dinge wie auf ähnliche Weise verzerrt.

Tabelle 1., Definitionen von Halluzination, Illusion und Verzerrung .
Die Unterscheidung zwischen Halluzination, Illusion und Verzerrung ist nicht nur eine akademische Frage. Im Gegenteil, es wird angenommen, dass ihre phänomenologischen Unterschiede Unterschiede in den zugrunde liegenden Mechanismen widerspiegeln. Pathophysiologisch werden Wahrnehmungsverzerrungen strukturellen oder funktionellen Läsionen diskreter Teile des Wahrnehmungsnetzwerks zugeschrieben, wie z. B. Bereich V4 bei Hyperchromatopsie und V5 bei Akinetopsie (23)., Es gibt acht bekannte Gruppen der zugrunde liegenden Ätiologie, einschließlich Infektionskrankheiten (Enzephalitis), Läsionen des zentralen Nervensystems (ZNS) (Schlaganfall, Hirntumor), Läsionen des peripheren Nervensystems (PNS) (Augenkrankheit, Mittelohrerkrankung), paroxysmale neurologische Störungen (Epilepsie, Migräne), psychiatrische Störungen (Depression, Schizophrenie), Medikamente, illegale Substanzen und eine andere Gruppe, zu der Hypnagogie und sensorische Deprivation gehören (4)., So rechtfertigen sogenannte „klinische Fälle“ von AIWS immer eine diagnostische Aufarbeitung, einschließlich neurologischer und psychiatrischer Beratung, Kopf-MRT und EEG und, falls angezeigt, anderer Hilfsuntersuchungen (z. B. ophthalmologische Untersuchung, HNO-Beratung). Obwohl evidenzbasierte Behandlungen noch entwickelt werden müssen, ist es eine gute klinische Praxis, die Therapie auf die (wahrscheinliche) zugrunde liegende Ursache auszurichten (4, 19).
Creutzfeldt-Jakob-Krankheit
Nach unserem Wissen wurde AIWS im Zusammenhang mit CJD noch nicht gemeldet. Mit 1-1.,5 Fälle pro Million Einwohner pro Jahr, CJD ist eine äußerst seltene Krankheit, die bei etwa 90% der Betroffenen innerhalb eines Jahres tödlich verläuft (24). Die Spitzeninzidenz liegt im siebten Jahrzehnt, während jüngere (20-40 Jahre) oder ältere (>80 Jahre) Fälle viel seltener sind (25). CJD gehört zu den übertragbaren spongiformen Enzephalopathien oder Prionenkrankheiten. Prionenkrankheiten sind durch die Ablagerung einer abnormal fehlgefalteten Isoform des nativen Prionproteins gekennzeichnet, die vom Prion-Gen (PRNP) auf dem menschlichen Chromosom 20 kodiert wird., Der Mechanismus zum Auslösen dieser Konformationsänderung ist unbekannt, aber die Akkumulation dieses abnormalen Prionenproteins führt zu neuronaler Degeneration, astrozytischer Gliose und spongiformen Veränderungen, begleitet von einem schnellen kognitiven Rückgang und endet mit dem Tod.,(ii) genetische CJD, die 10-15% der Fälle ausmacht und mit Mutationen des Prion-Gens (PRNP) assoziiert ist; (iii) die iatrogene Form von CJD, die etwa 1% der Fälle ausmacht und am häufigsten mit der vorherigen Behandlung mit humanen Hypophysen-abgeleiteten Hormonen oder menschlichen Dura-Mater-Transplantaten in Verbindung gebracht wird; und (iv) Variante CJD (vCJD), die eine neuartige humane Prion-Krankheit ist, die fast ausschließlich im Vereinigten Königreich vorkommt und mit dem Verzehr von Rindfleischprodukten in Verbindung gebracht wurde.mit dem Erreger der bovinen spongiformen Enzephalopathie (BSE) oder „Rinderwahnsinn.,“Darüber hinaus wurden basierend auf der anfänglichen klinischen Präsentation sechs Phänotypen identifiziert, die als kognitive, visuelle (Heidenhain-Variante), affektive, klassische, ataktische (Oppenheimer-Brownell-Variante) und unbestimmte (26) bezeichnet werden. Die Heidenhain-Variante wurde erstmals in den 1920er Jahren beschrieben und nach dem deutschen Physiologen und Histologen Rudolf Heidenhain (1834-1897) benannt., Es repräsentiert etwa 20% aller Fälle von sporadischer CJD (26-28) und ist durch isolierte visuelle Symptome zu Beginn der Erkrankung gekennzeichnet, wie Gesichtsfelddefekte und Verlust der Sehschärfe, die als Reflexionen einer frühen Beteiligung der Okzipitalrinde angesehen werden (29). Da diese Sehfehler kognitiven und motorischen Symptomen um mehrere Wochen vorausgehen können, ist es nicht ungewöhnlich, dass CJD in solchen Fällen nicht einmal vermutet wird und dass Patienten zunächst an einen Augenarzt überwiesen werden.,
Schlussfolgerung
Wir kommen zu dem Schluss, dass die Möglichkeit einer Heidenhain-Variante von CJD, obwohl äußerst selten, in der Differentialdiagnose von AIWS unterhalten werden muss, insbesondere bei schnellem kognitivem Rückgang. Darüber hinaus kommen wir zu dem Schluss, dass AIWS in den meisten Fällen relativ harmlos sein können, dass „klinische“ Fälle jedoch immer eine ordnungsgemäße diagnostische Aufarbeitung erfordern, bevor diese Schlussfolgerung gezogen werden kann.
Einschränkungen
Unser Wissen über die zahlreichen Ursachen und Erscheinungsformen von AIWS steckt noch in den Kinderschuhen., Das Syndrom wurde erstmals 1955 konzipiert, aber nur in den letzten Jahren ist die Anzahl der Veröffentlichungen zu dieser unterschiedlichen Gruppe von Phänomenen stetig gestiegen. Da das Gebiet noch sehr im Fluss ist, müssen unsere theoretischen Überlegungen zu AIWS, obwohl sie auf dem neuesten Stand der Technik sind, als vorläufig betrachtet werden. Zweitens bedeutet die Tatsache, dass wir hier den ersten Fallbericht über AIWS im Zusammenhang mit CJD präsentieren, nicht unbedingt, dass unser Patient die erste Person war, die jemals an diesen beiden seltenen Erkrankungen litt., Insbesondere kann erwartet werden, dass die Heidenhain-Variante von CJD häufiger mit Metamorphopsien auftritt. Daher kann unsere Unfähigkeit, frühere Fallbeschreibungen von Symptomen von AIWS im Zusammenhang mit CJD zu finden, eher auf eine Unterberichterstattung als auf eine tatsächliche Seltenheit der Kombination zurückzuführen sein. Eine dritte und letzte Einschränkung besteht darin, dass wir keine Zustimmung zur Autopsie erhalten konnten, was für die Diagnose einer bestimmten CJD erforderlich gewesen wäre.
Ethikerklärung
Die schriftliche Zustimmung zur Veröffentlichung wurde von der Familie des Patienten eingeholt.,
Beitrag zur Feldaussage
Das Alice-in-Wonderland-Syndrom (AIWS) ist eine seltene neurologische Störung, die durch Verzerrungen der visuellen Wahrnehmung, des Körperbildes und der Erfahrung der Zeit gekennzeichnet ist. Menschen können Dinge sehen, die kleiner sind als sie, fühlen, wie sich ihr Körper verändert oder erleben eines der zahlreichen anderen Symptome des Syndroms. Da es auch viele bekannte Ursachen für AIWS gibt, erfordert die Diagnose eine gründliche neurologische Aufarbeitung. Bei Kindern ist die häufigste Ursache eine Hirnentzündung, bei Erwachsenen ist es Migräne., In der medizinischen Literatur wurden nicht mehr als 180 einzelne Patienten beschrieben, von denen sich 50% erholten. Groß Angelegte Bevölkerungsstudien zeigen jedoch, dass flüchtige, vorübergehende Symptome von AIWS bei bis zu 30% aller gesunden Personen auftreten. Dementsprechend wird AIWS in der Regel als ziemlich harmlos angesehen. Wir stellen hier die erste Fallbeschreibung eines älteren Mannes vor, der aufgrund der Creutzfeldt-Jakob-Krankheit (CJD), einer äußerst seltenen neurodegenerativen Störung, an AIWS litt., Unser Patient entwickelte schnell einen Demenzzustand und starb innerhalb von 2 Monaten, nachdem seine visuellen Symptome begonnen hatten. Wir kommen zu dem Schluss, dass AIWS nicht immer harmlos ist und dass CJD, obwohl äußerst selten, den Ärzten in den Sinn kommen muss, wenn die für AIWS typischen visuellen Symptome von einem schnellen kognitiven Rückgang begleitet werden.,
Autorenbeiträge
TN trug zur Konzeption und Gestaltung der Arbeit sowie zur Erfassung, Analyse und Interpretation von Daten für die Arbeit bei, entwarf und überarbeitete die Arbeit, genehmigte die endgültige Veröffentlichung der endgültigen Version und erklärte sich bereit, für alle Aspekte der Arbeit verantwortlich zu sein, um sicherzustellen, dass Fragen im Zusammenhang mit der Genauigkeit oder Integrität eines Teils der Arbeit angemessen untersucht und gelöst werden., BtM und SvdW trugen zur Konzeption und Gestaltung der Arbeit sowie zur Erfassung, Analyse und Interpretation von Daten für die Arbeit bei, überarbeiteten die Arbeit, genehmigten die endgültige Veröffentlichung der endgültigen Version und erklärten sich bereit, für alle Aspekte der Arbeit verantwortlich zu sein, um sicherzustellen, dass Fragen im Zusammenhang mit der Genauigkeit oder Integrität eines Teils der Arbeit angemessen untersucht und gelöst werden., JDB trug zur Konzeption und Gestaltung der Arbeit sowie zur Analyse und Interpretation der Daten für die Arbeit bei, entwarf und überarbeitete die Arbeit, genehmigte die endgültige Veröffentlichung der endgültigen Version und erklärte sich bereit, für alle Aspekte der Arbeit verantwortlich zu sein, um sicherzustellen, dass Fragen im Zusammenhang mit der Genauigkeit oder Integrität eines Teils der Arbeit angemessen untersucht und gelöst werden.,
Interessenkonflikterklärung
Die Autoren erklären, dass die Untersuchung ohne kommerzielle oder finanzielle Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
1. Todd J. Das Syndrom von Alice im Wunderland. Can Med Assoc J. (1955) 73:701-4.
PubMed Abstract | Google Scholar
2. American Psychiatric Association. Diagnostisches und statistisches Handbuch für psychische Störungen. 5.Aufl., Washington, DC: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
CrossRef Full Text | Google Scholar
3. World Health Organization. The International Classification of Diseases, 10th Revision. Geneva: World Health Organization (1992).
Google Scholar
5. Abe K, Suzuki T., Prävalenz einiger Symptome in der Adoleszenz und Reife: Soziale Phobien, Angstsymptome, episodische Illusionen und Bezugsidee. Psychopathologie. (1986) 19:200–5. doi: 10.1159/000284448
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Lanska JR, Lanska DJ. Alice im Wunderland-Syndrom: somästhetische vs visuelle Wahrnehmungsstörung. Neurologie. (2013) 80:1262–4. doi: 10.1212/WNL.,0b013e31828970ae
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Bender MB, Kanzer MG. Metamorphopsie und andere psychovisuelle Störungen bei einem Patienten mit Tumor des Gehirns. Arch Neurol Psychiatrie. (1941) 45:481–5. doi: 10.1001/archneurpsyc.1941.02280150089005
CrossRef Volltext / Google Scholar
13. Carroll L. Alices Abenteuer im Wunderland. London: MacMillan (1865).,
Google Scholar
16. Podoll K, Robinson D. Migraine Art. The Migraine Experience From Within. Berkeley, CA: North Atlantic Books (2008).
Google Scholar
18. Carmichael C. Wonderland revisited. London Miscellany. (1996) 28:19–28.
Google Scholar
19. Blom JD. Alice in Wonderland Syndrome., Heidelberg: Springer (2019).
Google Scholar
22. Funatsu N, Hayakawa M, Tokuda N, Toyoda K. Vorübergehende Prosopometamorphopsie, die auf das linke Auge beschränkt ist und durch Ischämie am rechten Teil des Corpus callosum verursacht wird. Intern Med. (2017) 56:2933–5. doi: 10.2169/internalmedicine.8295-16
PubMed Abstract | CrossRef Volltext | Google Scholar
24., Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol. (2009) 66:208–15. doi: 10.1001/archneurol.2008.533
CrossRef Full Text | Google Scholar
26. Heidenhain A. Klinische und anatomische Untersuchungen über eine eigenartige organische Erkrankung des Zentralnervensystems im Praesenium. Z Ges Neurol Psychiat. (1929) 118:49–114. doi: 10.,1007/BF02892896
CrossRef Full Text | Google Scholar
28. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, et al. Revisiting the Heidenhain variant of Creutzfeldt-Jakob disease: evidence for prion type variabiliy influencing clinical course and laboratory findings. J Alzheimers Dis. (2016) 50:465–76. doi: 10.3233/JAD-150668
CrossRef Full Text | Google Scholar
29., Cooper SA, Murray KL, Heath CA, Wird RG, Ritter RSG. Isolierte visuelle Symptome zu Beginn der sporadischen Creutzfeldt-Jakob-Krankheit: der klinische Phänotyp der Heidenhain-Variante.“Br J Ophthalmol. (2005) 89:1341–2. doi: 10.1136/bjo.2005.074856
PubMed Abstract / CrossRef Volltext / Google Scholar