Sfondo
la sindrome di Alice nel paese delle Meraviglie (AIWS) è una rara malattia neurologica caratterizzata da distorsioni della percezione visiva (metamorphopsias), l’immagine del corpo e l’esperienza del tempo. Come notato già nel 1955 da John Todd, questi sintomi possono essere accompagnati da derealizzazione e depersonalizzazione (1)., I pazienti affetti da AIWS possono finire per consultare un neurologo o uno psichiatra, anche se in entrambe le specialità non è così noto come merita di essere. Ciò è dovuto almeno in parte al fatto che le principali classificazioni come il Manuale diagnostico e statistico dei disturbi mentali non lo elencano come categoria diagnostica, mentre altri, come la Classificazione internazionale delle malattie, prestano solo un’attenzione limitata ad esso. Un altro motivo potrebbe essere il numero relativamente piccolo di casi pubblicati., Una revisione della letteratura esistente, pubblicata nel 2016, ha indicato che solo 169 casi di AIWS erano stati descritti dalla concettualizzazione della sindrome nel 1955, che si riduce a un numero medio di 1,1 casi all’anno (4). Sebbene questo numero sia aumentato costantemente negli ultimi anni, attualmente si trova intorno a 180. Da notare, questi sono tutti i casi che necessitano di cure mediche, cioè quelli che chiamiamo “casi clinici.”Nel frattempo, studi sulla popolazione su larga scala indicano che i sintomi di AIWS sono sperimentati abbastanza frequentemente nella popolazione generale, con tassi di prevalenza fino al 30% (5-7)., Poiché questi ultimi casi sono in genere fugaci e transitori in natura, e raramente comportano un comportamento di ricerca di aiuto, essi sono indicati come “casi non clinici.”Dei casi clinici, circa l’ 85% mostra il coinvolgimento di una sola modalità sensoriale. Di questi, la maggior parte è limitata a un singolo sintomo (ad esempio, solo micropsia o solo macrosomatognosia, ecc.) (8). AIWS ha molte eziologie diverse. Nei casi clinici, l’encefalite (in particolare a causa dell’infezione da virus di Epstein-Barr) è la causa più comune nei bambini; negli adulti, è l’emicrania (4)., Sebbene la prognosi e l’esito dipendano in gran parte dal disturbo sottostante e dalla sua suscettibilità al trattamento, l’onere causato dall’AIWS può essere sostanziale, anche quando la sua causa è abbastanza innocua, come nel contesto della febbre o dell’uso sporadico di cannabis. Ciò è particolarmente vero quando i sintomi disturbano gravemente la percezione sensoriale, interferiscono con il proprio funzionamento quotidiano e / o evocano pensieri catastrofici (ad esempio, che il mondo è effettivamente cambiato in modo incomprensibile o che i sintomi possono essere indicativi di demenza o schizofrenia)., Poiché i casi non clinici sono per definizione autolimitanti, e anche nel 50% di tutti i casi clinici, la rassicurazione sembra essere sufficiente, l’AIWS è talvolta caratterizzato come relativamente innocuo in natura (9). Tuttavia, poiché la ricerca in questo settore è ancora agli inizi e l’AIWS potrebbe essere gravemente sottovalutata, è troppo presto per trarre conclusioni sulla sua storia naturale e sul risultato “tipico”. Inoltre, sappiamo già che l’AIWS può essere una manifestazione di condizioni gravi e persino letali, come infarto cerebrale o tumore al cervello (10, 11)., Presentiamo qui un caso di AIWS causato da sporadica malattia di Creutzfeldt-Jakob (CJD). Con esso, aggiungiamo alla letteratura fiorente su AIWS e cerchiamo di contrastare qualsiasi conclusione prematura che questa sindrome possa a priori essere caratterizzata come relativamente “benigna” in natura.
Metodi
Il paziente che descriviamo era in trattamento nella nostra pratica clinica. Dal momento che è morto entro 2 mesi dopo i suoi sintomi di AIWS avevano cominciato, abbiamo ottenuto il consenso scritto di pubblicare dalla sua famiglia.,
Risultati-Case Report
A marzo 2018, un uomo di 68 anni ha riferito alla nostra clinica ambulatoriale di neurologia con sintomi visivi. Ha avuto una storia di chirurgia della cataratta nel 2004 e sordità percezione a partire dal 2006. Poche settimane prima di vederlo, era stato indirizzato all’oftalmologo a causa della scarsa percezione della profondità e delle dimensioni e di una sensazione stretta intorno alla testa che era persistita per 1 mese. Non aveva altri sintomi (neurologici). La valutazione oftalmologica ha indicato un glaucoma ad angolo aperto con pressioni intraoculari di 39/27 mmHg ODS (riferimento 12-21 mmHg)., La sua acuità visiva era quasi normale (OD 0·8 dpt, OS 0·9 dpt), anche se l’esame del campo visivo ha rivelato diversi difetti lievi (OD) che non erano coerenti con i suoi sintomi. La sua pressione oculare normalizzata con l’uso di bimatoprost e dorzolamide/timololo collirio. Tuttavia, nel corso delle settimane successive, i sintomi visivi sono diventati più pronunciati e peculiari. Dopo il rinvio alla nostra clinica ambulatoriale, ha riferito che gli oggetti in movimento stavano rallentando e accelerando “come in un film di Charlie Chaplin” (noto come durata prolungata e fenomeno di movimento rapido, rispettivamente, cioè,, varianti del gruppo di distorsioni temporali). A volte non riusciva nemmeno a vedere alcun movimento (akinetopsia). Inoltre, ha percepito i colori come eccezionalmente brillanti (ipercromatopsia) e cambiando in modo casuale, come una maglietta rossa che diventa verde “come luci da fiera” (chloropsia/discromatopsia). Vide anche oggetti che si restringevano o si gonfiavano fino a dimensioni innaturali, “come se stesse guardando in uno specchio funhouse” (micropsia e macropsia, zoom vision)., Oltre a questi tipi di metamorfopsia, ha sviluppato parestesie nelle mani e nei piedi, instabilità dell’andatura e disfunzioni di concentrazione e memoria. Aveva anche difficoltà a suonare il flauto jazz e il sassofono, che consideravamo un segno inquietante, dato che era un musicista jazz professionista. Il suo discorso è diventato meno fluente perché non riusciva a trovare le parole giuste o non è riuscito a stringerle insieme per fare frasi, e il suo partner ha notato cambiamenti comportamentali sotto forma di agitazione e labilità emotiva.,
Sebbene l’esame neurologico, l’esame del sangue di routine e una risonanza magnetica della testa (inclusa l’imaging ponderato per la diffusione, DWI) non abbiano mostrato anomalie, il nostro paziente è diventato sempre più smemorato e disorientato e successivamente è stato ricoverato nel nostro ospedale. Lì abbiamo visto un uomo precedentemente eloquente che ora soffriva di afasia, atassia, cretini mioclonici e una demenza rapidamente progressiva., Una seconda risonanza magnetica ha mostrato valori di basso coefficiente di diffusione apparente (ADC) e un aumento della segnalazione DWI nella corteccia parieto-occipitale sinistra, caratteristica di un segno a nastro corticale (Figura 1), mentre l’elettroencefalogramma ha mostrato complessi periodici di onde taglienti (PSWC) sulla stessa area (Figura 2). La risonanza magnetica con presenza di proteine 14-3-3 e proteine tau elevate nel liquido cerebrospinale ha supportato la diagnosi di probabile malattia sporadica di Creutzfeldt-Jakob (CJD), in particolare la variante di Heidenhain., Abbiamo escluso altre possibili cause, tra cui disturbi autoimmuni, infezioni, tumori maligni, encefalopatie tossiche e metaboliche e altre malattie neurodegenerative (in particolare atrofia corticale posteriore e demenza con corpi di Lewy). Non abbiamo effettuato test genetici perché la storia familiare era negativa per la malattia neurodegenerativa, in particolare la CJD. Sfortunatamente non c’erano opzioni terapeutiche. In conformità con i suoi desideri, il nostro paziente è stato dimesso a casa, dove è morto entro 2 mesi dall’inizio dei suoi sintomi visivi. Non siamo stati in grado di ottenere il consenso dell’autopsia.,
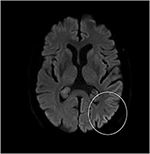
Figura 1. Alta intensità del segnale della corteccia parieto-occipitale sinistra su risonanza magnetica ponderata per diffusione (DWI; b = 1.000).
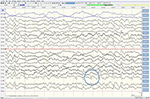
Figura 2. EEG che mostra complessi periodici di onde taglienti sulla regione parieto-occipitale sinistra, indicativi di CJD.,
Discussione
Ciò che rende questo caso unico, è che il nostro paziente ha presentato un numero relativamente elevato di metamorfopsias (che, nella letteratura esistente, è riportato solo nel 15% di tutti i casi clinici di AIWS) e che CJD è stata la causa più probabile. Dal momento che una diagnosi di CJD definita può essere stabilita solo post mortem mediante autopsia, è stato un peccato che non siamo stati in grado di ottenere il consenso dell’autopsia. Tuttavia, i risultati della risonanza magnetica sono risultati positivi nell ‘ 83% dei casi di probabile CJD (rispetto al 98% dei casi di CJD definita) (12)., Pertanto, anche se la risonanza magnetica della testa non è il gold standard, una diagnosi di CJD era ancora molto probabile nel nostro paziente, specialmente considerando il nostro ulteriore lavoro neurologico, che non è riuscito a produrre una diagnosi alternativa che potrebbe spiegare il quadro clinico, incluso il suo rapido declino e l’esito drammatico.,
Sindrome di Alice nel Paese delle Meraviglie
Come descritto da John Todd nel 1955, AIWS è caratterizzato da distorsioni percettive che ricordano le distorsioni visive, le distorsioni temporali, i cambiamenti corporei, la derealizzazione e la spersonalizzazione vissute da Alice durante il suo soggiorno nel Paese delle Meraviglie, come descritto nel classico libro per bambini di Lewis Carroll Alice’s Adventures in Wonderland (1, 13)., Todd, e prima di lui, Lipmann (14), suggerì che Carroll-il cui vero nome era Charles Lutwidge Dodgson-aveva trovato ispirazione per questi fenomeni peculiari nelle distorsioni percettive che aveva sperimentato se stesso nel contesto dell’emicrania. Sebbene questa ipotesi sia stata seguita da diversi trattati storici che asserivano che l’emicrania fosse effettivamente la fonte più probabile delle esperienze di Dodgson (15, 16), altri hanno avanzato l’ipotesi altrettanto allettante che, nel suo caso, fosse piuttosto l’epilessia a causarle (17) o forse l’abuso di sostanze (18)., Anche se probabilmente non sapremo mai con certezza cosa ha fatto Dodgson sperimentare questi sintomi, un’attenta ricostruzione della sua storia medica indica che la loro fonte più probabile era la malattia infettiva, di cui l’autore ha sofferto in numerose occasioni dall’infanzia in poi, letteralmente fino all’ultimo giorno della sua vita (19)., È molto probabile che Dodgson avesse sperimentato personalmente queste distorsioni (piuttosto che aver basato le esperienze della sua eroina su informazioni di terza mano), con un enorme 13 diversi sintomi descritti in tutto il libro, molto prima che Todd avesse inventato il concetto di AIWS.,
Per capire cosa distingue l’AIWS da altre sindromi e disturbi percettivi, dobbiamo renderci conto che le distorsioni differiscono dalle allucinazioni e dalle illusioni in quanto non sono né percezioni di qualcosa che non c’è (allucinazione), né oggetti o scene reali erroneamente giudicati come qualcos’altro (illusione). Invece, le distorsioni sono percezioni, sperimentate da un individuo sveglio, che si basano su stimoli appropriati dal mondo esterno, di cui un aspetto altamente specifico è alterato in modo coerente ., Oggi sono noti circa 55 diversi tipi di distorsione percettiva che possono verificarsi nel contesto dell’AIWS (4). Quelli più frequentemente riportati sono micropsia (vedere le cose più piccole di quanto non siano), macropsia (vedere le cose più grandi di quanto non siano), teleopsia (vedere le cose più lontane di quanto non siano) e dismorfopsia (vedere le linee rette come ondulate). La disposizione che tali aspetti sono alterati in modo coerente, significa che la distorsione si applica a qualsiasi cosa all’interno del raggio visivo del paziente., Quindi, le persone che soffrono di plagiopsia vedranno tipicamente tutte le cose come inclinate, nella stessa direzione e sotto lo stesso grado, mentre quelle con prosopometamorfopsia vedranno tipicamente una particolare alterazione in tutti i volti che percepiscono, come sopracciglia pesanti o un occhio che è marcatamente rapito al naso o volti umani che si trasformano in volti di drago (21, 22). La natura di queste distorsioni può variare nel tempo, ma una volta presenti, tutte le cose percepite appaiono come distorte in modo simile.

Tabella 1., Definizioni di allucinazione, illusione e distorsione .
La distinzione tra allucinazione, illusione e distorsione non è solo una questione accademica. Al contrario, si pensa che le loro differenze fenomenologiche riflettano differenze nei meccanismi sottostanti. Fisiopatologicamente, le distorsioni percettive sono attribuite a lesioni strutturali o funzionali di parti discrete della rete percettiva, come l’area V4 nell’ipercromatopsia e V5 nell’acinetopsia (23)., Ci sono otto gruppi di eziologia sottostante, composto da malattie infettive (encefalite), del sistema nervoso centrale (SNC) lesioni (ictus, tumore al cervello, sistema nervoso periferico (PNS) lesioni (malattia dell’occhio, medio-malattia dell’orecchio), parossistica, disturbi neurologici (epilessia, emicrania), disturbi psichiatrici (depressione, schizofrenia), farmaci, sostanze illecite, e una miscellanea di un gruppo che comprende hypnagogia e deprivazione sensoriale (4)., Pertanto, i cosiddetti “casi clinici” di AIWS giustificano sempre il lavoro diagnostico, compresa la consultazione neurologica e psichiatrica, la risonanza magnetica della testa e l’EEG e, quando indicato, altre indagini ausiliarie (ad esempio, esame oftalmologico, consultazione ORL). Sebbene i trattamenti basati sull’evidenza siano ancora da sviluppare, è buona pratica clinica mirare la terapia alla (probabile) causa sottostante (4, 19).
Malattia di Creutzfeldt-Jakob
A nostra conoscenza, AIWS non è stato segnalato prima nel contesto di CJD. Con 1-1.,5 casi per milione di abitanti all’anno, la CJD è una malattia estremamente rara che è fatale entro un anno in circa il 90% delle persone colpite (24). L’incidenza di picco è nella settima decade, mentre i casi più giovani (20-40 anni) o più anziani (>80 anni) sono molto meno comuni (25). La CJD appartiene alle encefalopatie spongiformi trasmissibili o alle malattie da prioni. Le malattie da prioni sono caratterizzate dalla deposizione di un’isoforma anomala della proteina prionica nativa, che è codificata dal gene prionico (PRNP) sul cromosoma umano 20., Il meccanismo per innescare questo cambiamento conformazionale è sconosciuto, ma l’accumulo di questa proteina prionica anormale porta alla degenerazione neuronale, gliosi astrocitica e cambiamenti spongiformi, accompagnati da un rapido declino cognitivo e terminanti con la morte.,ximately 85% dei casi; (ii) genetica del morbo di creutzfeldt jacob, che rappresenta il 10-15% dei casi è associata a mutazioni del gene del prione (PRNP); (iii) il iatrogena forma di morbo di creutzfeldt jacob, che rappresenta circa l ‘ 1% dei casi ed è più frequentemente associato a un precedente trattamento con pituitaria umana ormoni derivati o umana dura mater innesti; e (iv) variante del morbo di creutzfeldt jacob (vCJD), che è un romanzo umano malattia da prioni trova quasi esclusivamente nel regno UNITO, ed è stato collegato al consumo di carne di manzo prodotti contaminati con l’agente eziologico della encefalopatia spongiforme bovina (BSE) o “malattia della mucca pazza.,”Inoltre, sulla base della presentazione clinica iniziale, sono stati identificati sei fenotipi, chiamati cognitivi, visivi (variante di Heidenhain), affettivi, classici, atattici (variante di Oppenheimer-Brownell) e indeterminati (26). La variante di Heidenhain è stata descritta per la prima volta durante gli anni 1920 e prende il nome dal fisiologo e istologo tedesco Rudolf Heidenhain (1834-1897)., Rappresentando circa il 20% di tutti i casi di CJD sporadica (26-28), è caratterizzata da sintomi visivi isolati all’inizio della malattia, come difetti del campo visivo e perdita dell’acuità visiva, che sono considerati riflessi del coinvolgimento precoce della corteccia occipitale (29). Poiché questi difetti visivi possono precedere i sintomi cognitivi e motori di diverse settimane, non è raro che la CJD non sia nemmeno sospettata in questi casi e che i pazienti siano inizialmente indirizzati a un oftalmologo.,
Conclusione
Concludiamo che la possibilità di una variante Heidenhain di CJD, sebbene estremamente rara, deve essere intrattenuta nella diagnosi differenziale di AIWS, specialmente in presenza di un rapido declino cognitivo. Inoltre, concludiamo che l’AIWS può essere relativamente innocuo nella maggior parte dei casi, ma che i casi “clinici” giustificano sempre un adeguato lavoro diagnostico prima che questa conclusione possa essere tratta.
Limitazioni
La nostra conoscenza delle numerose cause e manifestazioni di AIWS è ancora agli inizi., La sindrome fu concettualizzata per la prima volta nel 1955, ma solo negli ultimi anni si è assistito a un costante aumento del numero di pubblicazioni su questo gruppo eterogeneo di fenomeni. Poiché l’area è ancora molto in evoluzione, le nostre riflessioni teoriche sull’AIWS, sebbene allo stato dell’arte, devono essere considerate di natura preliminare. In secondo luogo, il fatto che presentiamo qui il primo rapporto di caso su AIWS nel contesto di CJD non implica necessariamente che il nostro paziente sia stato la prima persona a soffrire di queste due rare condizioni., In particolare, ci si può aspettare che la variante Heidenhain di CJD presenti più frequentemente con metamorfopsias. Pertanto, la nostra incapacità di trovare qualsiasi precedente descrizione dei sintomi di AIWS nel contesto di CJD potrebbe essere dovuta alla sottorapportazione piuttosto che a una rarità effettiva della combinazione. Un terzo e ultimo limite è che non siamo stati in grado di ottenere il consenso dell’autopsia, che sarebbe stato necessario per fare una diagnosi di CJD definita.
Dichiarazione etica
Il consenso scritto alla pubblicazione è stato ottenuto dalla famiglia del paziente.,
Contributo alla Dichiarazione sul campo
La sindrome di Alice nel Paese delle meraviglie (AIWS) è una rara malattia neurologica caratterizzata da distorsioni della percezione visiva, dell’immagine corporea e dell’esperienza del tempo. Le persone possono vedere le cose più piccole di quanto non siano, sentire il loro corpo alterare le dimensioni o sperimentare uno qualsiasi dei numerosi altri sintomi della sindrome. Poiché ci sono anche molte cause note di AIWS, la diagnosi richiede un accurato lavoro neurologico. Nei bambini, la causa più comune è l’infiammazione cerebrale; negli adulti, è l’emicrania., Nella letteratura medica non sono stati descritti più di 180 singoli pazienti, il 50% dei quali ha recuperato. Tuttavia, studi sulla popolazione su larga scala indicano che i sintomi fugaci e transitori di AIWS sono sperimentati fino al 30% di tutti gli individui sani. Di conseguenza, AIWS è generalmente considerato abbastanza innocuo in natura. Presentiamo qui la prima descrizione del caso di un uomo anziano che soffriva di AIWS a causa della malattia di Creutzfeldt-Jakob (CJD), una malattia neurodegenerativa estremamente rara., Il nostro paziente sviluppò rapidamente uno stato di demenza e morì entro 2 mesi dall’inizio dei suoi sintomi visivi. Concludiamo che l’AIWS non è sempre innocuo e che la CJD, sebbene estremamente rara, deve essere nella mente dei medici quando i sintomi visivi tipici dell’AIWS sono accompagnati da un rapido declino cognitivo.,
Autore Contributi
TN contribuito alla ideazione e la progettazione di lavori e per l’acquisizione, l’analisi e l’interpretazione dei dati per il lavoro, redatto e revisionato il lavoro, ha dato l’approvazione finale per la versione definitiva, che dovrà essere pubblicato, e accettato di essere responsabile per tutti gli aspetti del lavoro nel garantire che le questioni relative all’accuratezza o l’integrità di qualsiasi parte del lavoro sono adeguatamente studiato e risolto., BtM e SvdW hanno contribuito alla concezione e alla progettazione dell’opera e all’acquisizione, analisi e interpretazione dei dati per l’opera, hanno rivisto l’opera, hanno dato l’approvazione finale per la versione finale da pubblicare e hanno accettato di essere responsabili di tutti gli aspetti dell’opera nel garantire che le questioni relative all’accuratezza o all’integrità di qualsiasi parte dell’opera siano adeguatamente investigate e risolte., JDB ha contribuito alla concezione e alla progettazione dell’opera, all’analisi e all’interpretazione dei dati per l’opera, ha redatto e rivisto l’opera, ha dato l’approvazione finale per la versione finale da pubblicare e ha accettato di essere responsabile di tutti gli aspetti dell’opera nel garantire che le questioni relative all’accuratezza o all’integrità di qualsiasi parte dell’opera siano adeguatamente investigate e risolte.,
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
1. Todd J. La sindrome di Alice nel paese delle Meraviglie. Può Med Assoc J. (1955) 73:701-4.
PubMed Abstract/Google Scholar
2. Associazione Psichiatrica Americana. Manuale Diagnostico e Statistico dei Disturbi Mentali. 5a ed., Washington, DC: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
CrossRef Full Text | Google Scholar
3. World Health Organization. The International Classification of Diseases, 10th Revision. Geneva: World Health Organization (1992).
Google Scholar
5. Abe K, Suzuki T., Prevalenza di alcuni sintomi nell’adolescenza e nella maturità: fobie sociali, sintomi d’ansia, illusioni episodiche e idea di riferimento. Psicopatologia. (1986) 19:200–5. doi: 10.1159 / 000284448
PubMed Abstract | CrossRef Full Text/Google Scholar
8. Lanska JR, Lanska DJ. Sindrome di Alice nel Paese delle Meraviglie: disturbo percettivo somestetico vs visivo. Neurologia. (2013) 80:1262–4. doi: 10.1212 / WNL.,0b013e31828970ae
PubMed Abstract | CrossRef Full Text/Google Scholar
11. Bender MB, Kanzer MG. Metamorfopsia e altri disturbi psicovisuali in un paziente con tumore al cervello. Arch Neurol Psichiatria. (1941) 45:481–5. doi: 10.1001 / archneurpsyc.1941.02280150089005
CrossRef Full Text/Google Scholar
13. Le avventure di Carroll L. Alice nel paese delle meraviglie. Londra: MacMillan (1865).,
Google Scholar
16. Podoll K, Robinson D. Migraine Art. The Migraine Experience From Within. Berkeley, CA: North Atlantic Books (2008).
Google Scholar
18. Carmichael C. Wonderland revisited. London Miscellany. (1996) 28:19–28.
Google Scholar
19. Blom JD. Alice in Wonderland Syndrome., Heidelberg: Springer (2019).
Google Scholar
22. Funatsu N, Hayakawa M, Tokuda N, Toyoda K. prosopometamorphopsia transitoria limitata all’occhio sinistro causata da ischemia allo splenio destro del corpo calloso. Stagista Med. (2017) 56:2933–5. doi: 10.2169 / internalmedicina.8295-16
PubMed Abstract | CrossRef Full Text/Google Scholar
24., Apple B, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Caratteristiche delle varianti sporadiche della malattia di Creutzfeldt-Jakob stabilite e proposte. Arch Neurol. (2009) 66:208-15. doi: 10.1001 / archneurol.2008.533
CrossRef Full Text/Google Scholar
26. Heidenhain A. Studi clinici e anatomici di una particolare malattia organica del sistema nervoso centrale nel Praesenium. Z Ges Neurol Psychiat. (1929) 118:49-114. doi: 10.,1007/BF02892896
CrossRef Full Text | Google Scholar
28. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, et al. Revisiting the Heidenhain variant of Creutzfeldt-Jakob disease: evidence for prion type variabiliy influencing clinical course and laboratory findings. J Alzheimers Dis. (2016) 50:465–76. doi: 10.3233/JAD-150668
CrossRef Full Text | Google Scholar
29., I nostri servizi sono a vostra disposizione. Sintomi visivi isolati all’esordio nella malattia sporadica di Creutzfeldt-Jakob: il fenotipo clinico della ” variante di Heidenhain.”Br J Ophthalmol. (2005) 89:1341–2. doi: 10.1136 / bjo.2005.074856
PubMed Abstract | CrossRef Full Text/Google Scholar