INTRODUZIONE
sindrome di Klippel-Trénaunay-Weber sindrome di (KTWS) è caratterizzata da un insieme di segni che consistono di malformazioni capillari, malformazioni venose con o senza malformazioni linfatiche associati con arto overgrowth1. Nella maggior parte dei casi, coinvolge solo un’estremità con malformazione artero-venosa e circa il 75% dei pazienti manifesta la malattia prima dei 10 anni di età2,3.,
La sindrome di Klippel-Trenaunay è stata descritta per la prima volta da Maurice Klippel e Paul Trenaunay. Hanno riportato due casi che avevano la triade (macchia di vino porto, varici e ipertrofia ossea e dei tessuti molli) in comune4. Qualche tempo dopo, Frederick Weber descrisse alcuni casi che presentavano una somiglianza con la triade,con la presenza di fistola artero-venosa come associazione3, 4. La sindrome di SKTW e Parkes Weber può essere considerata insieme come Klippel-Trenaunay-Weber sd perché presentano diversi segni clinici di una singola malattia5,6.,
KTWS è un raro disturbo mesodermico congenito, con circa 1000 casi registrati in tutto il mondo7. KTWS è distribuito equamente tra i vari gruppi etnici e colpisce più uomini, con un rapporto di 1.5:17,8. L’eziologia di questa sindrome rimane sconosciuta, sebbene ci siano alcune teorie sulla sua patogenesi9.
Sebbene KTWS sia una condizione sporadica, gli studi hanno riportato casi familiari di KTWS che non sono stati ereditati attraverso un modello mendeliano, suggerendo un’eredità multifattoriale, con penetrazione ed espressione variabili., Successivi studi condotti da Happle suggeriscono che l’ereditarietà di un singolo gene difettoso acquisito durante l’embriogenesi potrebbe spiegare lo sviluppo di questa sindrome, così come l’insorgenza di casi sporadici e familiari, suggerendo che un’eredità autosomica dominante è più probabile10,11.
Clinicamente, KTWS comprende emangioma piatto, alterazioni venose come malformazioni e vene varicose e ipertrofia ossea e dei tessuti molli. 12,13
La diagnosi è clinica e può essere eseguita dalla presenza della triade di anomalie o solo due segni della triade.,
Di solito, i pazienti con macchie di vino porto, dalla nascita, principalmente negli arti ipertrofici, variando in profondità14. Le malformazioni venose colpiscono gli arti inferiori nella stragrande maggioranza dei casi. L’angiodisplasia arteriosa o venosa può essere presente in qualsiasi regione del corpo, dalla pelle agli organi viscerali. Pertanto, vi è la possibilità di flebiti, sanguinamento, trombosi venosa profonda, embolia polmonare, emoperitoneo, emotorace e insufficienza venosa cronica15.
È una sindrome rara, ma merita di essere evidenziata a causa della morbilità progressiva e grave.,
CASE REPORT
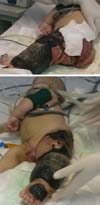
D. A. P., 7 mesi, maschio, nato e residente ad Anápolis-GO, è stato inoltrato al nostro servizio nel Pronto soccorso dell’Hospital de Clínicas, Università Federale di Uberlândia, MG, il 29/12/2014 con febbre, vomito, diarrea e disidratazione per 2 giorni. Insieme a questa presentazione, ha presentato un emangioma gigante nell’arto inferiore sinistro, vene varicose e ipertrofia delle ossa e dei tessuti molli (KTWS) (Figura 1) con una storia di recente trattamento laser in un altro servizio di Goiânia-GO e con propranololo.,

Il paziente progrediva rapidamente e gravemente con necrosi dell’arto inferiore sinistro (LLL) fino alla regione glutea ipsilaterale e con shock settico refrattario (Figura 2). Ha ricevuto un trattamento intensivo di terapia intensiva con farmaci vasopressori e terapia antibiotica ad ampio spettro per 28 giorni. L’eco Doppler arterioso e venoso è stato eseguito durante la valutazione della chirurgia vascolare, confermando la diagnosi della sindrome di Klippel-Trénaunay-Weber e identificando malformazioni artero-venose e piccole fistole dell’arto sinistro., Tuttavia, un’estesa lesione necrotica nella LLL è rimasta, richiedendo, principalmente, lo sbrigliamento chirurgico della LLL e successivamente la disarticolazione dell’anca sinistra con colostomia protettiva il 28/01/2015.
Dopo 2 mesi di trattamento clinico intensivo, è stato osservato un miglioramento clinico e topico della lesione nella regione postoperatoria della disarticolazione(Figura 3A) e l’innesto cutaneo è stato eseguito con dermatoma elettrico e innesto di poltiglia per l’occlusione della ferita sanguinante (Figura 3B), con l’area donatrice della coscia destra. Il paziente ha presentato un buon recupero clinico e l’epitelizzazione dell’area ricevente e donatrice, venendo dimesso il 15 ° giorno postoperatorio(Figura 3C).,

Il paziente è stato in follow up ambulatoriale in questo servizio, progredendo in modo soddisfacente con la completa epitelizzazione delle aree donatrice e ricevente. La colostomia protettiva è stata chiusa con la ricostruzione del transito intestinale.
DISCUSSIONE
Questo studio è stato motivato dall’aver ricevuto un paziente che si riferiva a un trattamento precedente in un altro servizio di chirurgia plastica con trattamento conservativo di KTWS con laser., Tale condotta ha mantenuto il paziente incapace, soffrendo di sintomi di stasi venosa cronica e si è evoluto con complicazioni di infezione cutanea locale fino a ricevere il trattamento finale.
Non esiste un trattamento curativo e gli obiettivi terapeutici hanno lo scopo di migliorare i sintomi del paziente e correggere le conseguenze di lesioni gravi e discrepanza di lunghezza. Tuttavia, tutti gli autori concordano sul fatto che le misure conservative continuano a guidare il trattamento di KTWS. Ciò non esclude la necessità di interventi chirurgici tempestivi durante l’evoluzione della storia naturale della malattia.,
Le terapie adiuvanti possono variare dalla terapia laser, con scleroterapia microfoam, resezioni in scena di vene ectatiche e escisioni ancora più grandi1, 16-19 . Le indicazioni più comunemente utilizzate per il trattamento chirurgico sono: emorragia, infezioni locali, tromboembolia e comparsa di ulcere alle gambe molto refrattarie. Altre indicazioni sono: dolore locale, limitazioni funzionali ed estetiche17.
La radioterapia interventistica svolge un ruolo importante nella propedeutica delle malformazioni artero-venose (AVMs)., Attraverso di esso, si può valutare il tipo di malformazione e come sono strutturati i vasi di alimentazione. Per il trattamento di AVM a basso flusso (KTWS), in alcuni casi può essere applicata un’iniezione di agente sclerosante per ridurre i vasi.
In altri casi, questo può essere fatto usando la fluoroscopia. Ci sono opzioni limitate per il trattamento della displasia venosa congenita. Nei casi più gravi, l’interventista può utilizzare la rimozione chirurgica, la scleroterapia o una tecnica di ablazione endovascolare. Se ci sono sintomi nella pelle, come macchie di vino porto, può essere indicato un trattamento laser.,
In casi come quello descritto, considerando il trattamento eseguito, l’area coperta dall’innesto può essere vista come una buona opzione ricostruttiva, tenendo conto della semplicità della procedura, della minore morbilità rispetto all’uso dei lembi e della possibilità di riabilitazione attraverso l’uso di protesi.,
Pur essendo il più alto livello di amputazione degli arti inferiori, la protesi è efficiente, poiché la protesi per questo livello di amputazione fornisce sicurezza e stabilità, con andatura continua, con o senza aiuti alla locomozione, a seconda di altri fattori tra cui l’età del paziente.
KTWS deve essere sospettato in tutti i neonati con malformazioni capillari che coinvolgono un’estremità del corpo dalla nascita. La diagnosi differenziale per KTWS è la sindrome di Proteus e la sindrome di Maffucci, tra le altre malformazioni capillari non sindromiche20.,
Grandi progressi saranno fatti quando sarà possibile diagnosticare KTWS ancora più presto e prevenire lo sviluppo di ipertrofia tissutale, angiodisplasie complesse e altri cambiamenti fenotipici, magari correggendo o prevenendo la relativa mutazione genetica20.
CONCLUSIONE
KTWS è una malattia rara, con morbilità progressiva e grave. Il paziente con questa sindrome deve essere accompagnato in un centro di riferimento con esperienza e un arsenale terapeutico diversificato per agire nel miglior modo possibile nel trattamento., Ogni paziente ha la parsimonia e l’individualizzazione nella scelta del trattamento migliore, così come il momento ideale per realizzarlo.
Attualmente, l’indicazione per l’intervento chirurgico è limitata alle complicanze derivanti dalla presentazione iniziale. In questo caso, l’uso del trattamento chirurgico è stato fondamentale per consentire un miglioramento delle condizioni cliniche e della qualità di vita del paziente, dimostrando che può essere una valida alternativa.,
COLLABORAZIONI
|
VPB |
Scrivere il manoscritto o la revisione critica del suo contenuto. |
|
AOM |
Analisi e / o interpretazione dei dati; scrittura del manoscritto o revisione critica del suo contenuto. |
2. Favorito LA. Emangioma vescicale in paziente con sindrome di Klippel-Trenaunay-Weber. J Urol. 2003;29(2):149-50.
4. Klippel M, Trénaunay P. Du naevus variqueux osteohypertrophique. Arch Gen Med (Parigi)., 1900;185:641-72.
5. Weber FP. Ipertrofia emangiectatica degli arti: flebarteriectasi congenita e cosiddette vene varicose congenite. Br J Bambino Dis. 1918;15:13-7.
9. Meine JG, Schwartz RA, Janniger CK. Sindrome di Klippel-Trenaunay-Weber. Cutis. 1997;60(3):127-32. PMID: 9314616
1. Universidade Federal de Uberlândia, Uberlândia, MG, Brasile.
Istituzione: Universidade Federal de Uberlândia, Uberlândia, MG, Brasile.