INTRODUCTION
le syndrome de Klippel-Trénaunay-Weber (KTWS) est caractérisé par un ensemble de signes qui consistent en des malformations capillaires, des malformations veineuses avec ou sans malformations lymphatiques associées à envahissement des membres1. Dans la plupart des cas, il ne s’agit que d’une extrémité présentant une malformation artérioveineuse et environ 75% des patients manifestent la maladie avant l’âge de 10 ans2,3.,
le syndrome de Klippel-Trenaunay a été décrit pour la première fois par Maurice Klippel et Paul Trenaunay. Ils ont signalé deux cas qui avaient la triade (tache de vin de Porto, varices et hypertrophie des os et des tissus mous) en commun4. Quelque temps plus tard, Frederick Weber a décrit certains cas présentant une similitude avec la triade,avec la présence d’une fistule artérioveineuse comme association3, 4. Le Syndrome de SKTW et le Syndrome de Parkes Weber peuvent être considérés ensemble comme sd de Klippel-Trenaunay-Weber car ils présentent des signes cliniques différents d’une même maladie5,6.,
Le KTWS est un trouble mésodermique congénital rare, avec environ 1000 cas enregistrés dans le monde7. Le KTWS est réparti également entre les différents groupes ethniques et touche davantage d’hommes, avec un ratio de 1,5:17,8. L’étiologie de ce syndrome reste inconnue, bien qu’il existe certaines théories de son pathogène9.
bien que le syndrome de KTWS soit une affection sporadique, des études ont rapporté des cas familiaux de syndrome de KTWS qui n’étaient pas hérités selon un modèle mendélien, suggérant un héritage multifactoriel, avec une pénétration et une expression variables., Des études ultérieures menées par Happle suggèrent que l’hérédité d’un seul gène défectueux acquis au cours de l’embryogenèse pourrait expliquer le développement de ce syndrome, ainsi que l’apparition de cas sporadiques et familiaux, suggérant qu’un héritage autosomique dominant est le plus probable10,11.
Cliniquement, KTWS comprend un hémangiome plat, des modifications veineuses telles que des malformations et des varices, et une hypertrophie osseuse et des tissus mous. 12,13
Le diagnostic est clinique et peut être effectuée par la présence de la triade des anomalies, ou seulement deux signes de la triade.,
habituellement, les patients atteints de taches de Porto, dès la naissance, principalement dans les membres hypertrophiés, variant en profondeur14. Les malformations veineuses affectent les membres inférieurs dans la grande majorité des cas. Une angiodysplasie artérielle ou veineuse peut être présente dans n’importe quelle région du corps, de la peau aux organes viscéraux. Par conséquent, il existe une possibilité de phlébite, de saignement, de thrombose veineuse profonde, d’embolie pulmonaire, d’hémopéritoine, d’hémothorax et d’insuffisance veineuse chronique15.
c’est un syndrome rare, mais mérite d’être mis en évidence en raison d’une morbidité progressive et sévère.,
rapport de cas
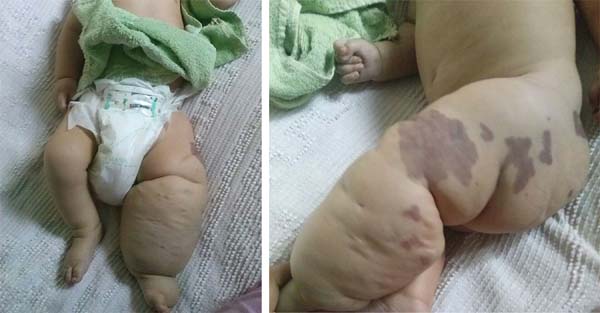
D. A. P., 7 mois, homme, né et résidant à Anápolis-GO, a été transmis à notre service dans la salle d’urgence de L’Hôpital de Clínicas, Université Fédérale D’Uberlândia, MG, le 29/12/2014 avec de la fièvre, des vomissements, de la diarrhée et de la déshydratation pendant 2 jours. Avec cette présentation, il a présenté un hémangiome géant dans le membre inférieur gauche, des varices et une hypertrophie des os et des tissus mous (Ktws) (Figure 1) avec une histoire de traitement au laser récent dans un autre service de Goiânia-GO et avec du propranolol.,

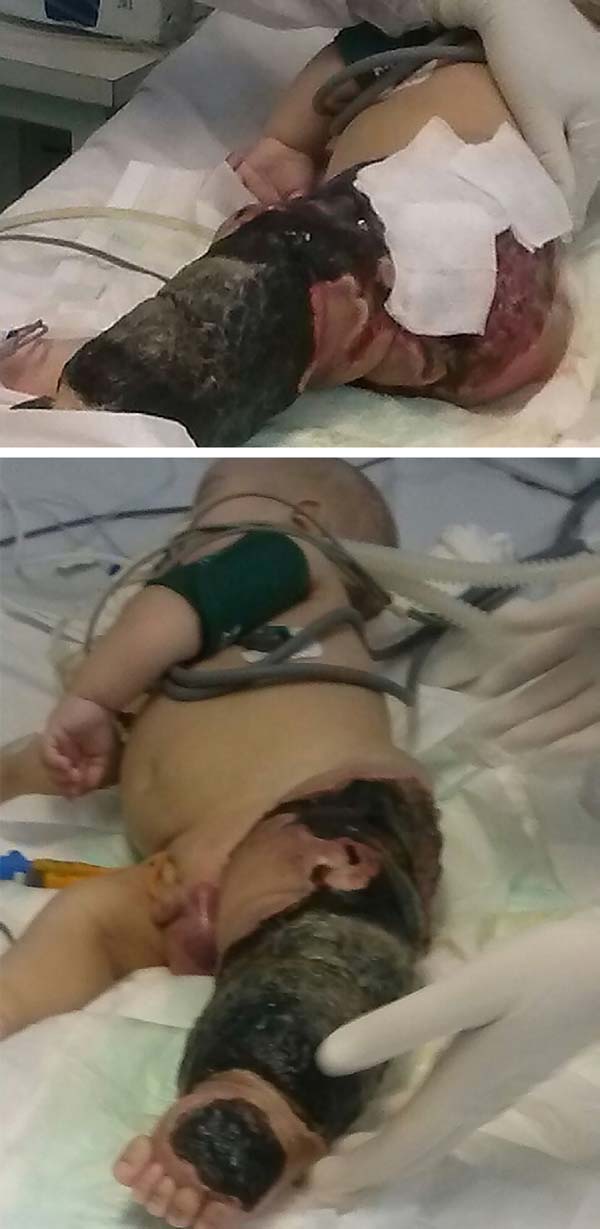
le patient a progressé rapidement et sévèrement avec une nécrose du membre inférieur gauche (LLL) jusqu’à la région fessière ipsilatérale et avec un choc septique réfractaire (Figure 2). Il a reçu un traitement intensif aux soins intensifs avec des médicaments vasopresseurs et une antibiothérapie à large spectre pendant 28 jours. Un écho-Doppler artériel et veineux a été réalisé lors de l’évaluation de la chirurgie vasculaire, confirmant le diagnostic du syndrome de Klippel-Trénaunay-Weber et identifiant une malformation artério-veineuse et de petites fistules du membre gauche., Cependant, une lésion nécrotique étendue de la LLL est restée, nécessitant principalement un débridement chirurgical de la LLL et par la suite une désarticulation de la hanche gauche avec colostomie protectrice le 28/01/2015.
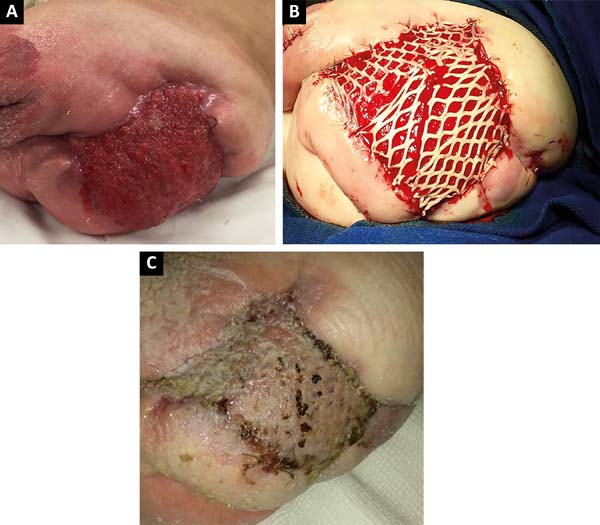
Après 2 mois de traitement clinique intensif, une amélioration clinique et topique de la lésion a été observée dans la région postopératoire de la désarticulation(Figure 3A), et une greffe de peau a été réalisée avec un dermatome électrique et une greffe de purée pour l’occlusion de la plaie sanglante (Figure 3B), avec la zone donneuse de Le patient a présenté une bonne récupération clinique et une épithélialisation de la zone du receveur et du donneur, étant libéré le 15e jour postopératoire(Figure 3C).,

le patient a été suivi ambulatoire dans ce service, progressant de manière satisfaisante avec une épithélialisation complète des zones du donneur et du receveur. La colostomie protectrice a été fermée avec la reconstruction du transit intestinal.
DISCUSSION
Cette étude a été motivée par le fait qu’un patient avait reçu un traitement antérieur dans un autre service de chirurgie plastique avec un traitement conservateur de KTWS au laser., Une telle conduite a maintenu le patient incapable, souffrant de symptômes de stase veineuse chronique et a évolué avec des complications d’infection cutanée locale jusqu’à la réception du traitement final.
Il n’y a pas de traitement curatif, et les objectifs thérapeutiques sont destinés à améliorer les symptômes du patient et à corriger les conséquences des blessures graves et des écarts de longueur. Cependant, tous les auteurs conviennent que des mesures conservatrices continuent de guider le traitement des TCA. Cela n’exclut pas la nécessité d’interventions chirurgicales en temps opportun au cours de l’évolution de l’histoire naturelle de la maladie.,
Les thérapies adjuvantes peuvent varier de la thérapie au laser, avec la sclérothérapie en microfoam, les résections étagées des veines ectatiques et les excisions encore plus larges1,16-19 . Les indications les plus couramment utilisées pour le traitement chirurgical sont: hémorragie, infections locales, thromboembolie et apparition d’ulcères de jambe très réfractaires. D’autres indications sont: douleur locale, limitations fonctionnelles et esthétiques17.
la radiothérapie interventionnelle joue un rôle majeur dans la propédeutique des malformations artério-veineuses (MAV)., Grâce à elle, on peut évaluer le type de malformation et la structure des vaisseaux d’alimentation. Pour le traitement des MAV à faible débit (KTWS), dans certains cas, une injection d’agent sclérosant peut être appliquée pour réduire les vaisseaux.
dans d’autres cas, cela peut être fait en utilisant la fluoroscopie. Il existe des options limitées pour le traitement de la dysplasie veineuse congénitale. Dans les cas graves, l’interventionniste peut utiliser l’ablation chirurgicale, la sclérothérapie ou une technique d’ablation endovasculaire. S’il y a des symptômes dans la peau, tels que des taches de vin de Porto, un traitement au laser peut être indiqué.,
dans des cas tels que celui décrit, compte tenu du traitement effectué, la zone couverte par la greffe peut être considérée comme une bonne option reconstructrice, compte tenu de la simplicité de la procédure, moins de morbidité par rapport à l’utilisation de volets et la possibilité de réhabilitation par l’utilisation de prothèses.,
en dépit d’être le plus haut niveau d’amputation des membres inférieurs, la prothèse est efficace, car la prothèse pour ce niveau d’amputation offre la sécurité et la stabilité, avec une démarche continue, avec ou sans aides à la locomotion, en fonction d’autres facteurs, y compris l’âge du patient.
Le KTWS doit être suspecté chez tous les nouveau-nés présentant des malformations capillaires impliquant une extrémité du corps dès la naissance. Le diagnostic différentiel de KTWS est le syndrome de Proteus et le syndrome de Maffucci, entre autres malformations capillaires non syndromiques20.,
des progrès majeurs seront réalisés lorsqu’il sera possible de diagnostiquer encore plus tôt les TCA et de prévenir le développement d’une hypertrophie tissulaire, d’angiodysplasies complexes et d’autres modifications phénotypiques, peut-être en corrigeant ou en prévenant la mutation génétique connexe 20.
CONCLUSION
Le KTWS est une maladie rare, avec une morbidité progressive et sévère. Le patient atteint de ce syndrome doit être accompagné dans un centre de référence avec de l’expérience et un arsenal thérapeutique diversifié pour agir de la meilleure façon possible dans le traitement., Chaque patient a la parcimonie et de l’individualisation dans le choix du meilleur traitement, ainsi que le moment idéal pour l’accomplir.
actuellement, l’indication d’une intervention chirurgicale est limitée aux complications résultant de la présentation initiale. Dans ce cas, l’utilisation du traitement chirurgical était cruciale pour permettre une amélioration de l’état clinique et de la qualité de vie du patient, montrant qu’il peut être une alternative valable.,
COLLABORATIONS
|
LISTE |
l’Écriture d’un manuscrit ou d’un examen critique de son contenu. |
|
CG |
l’Analyse et/ou de l’interprétation des données; l’écriture d’un manuscrit ou d’un examen critique de son contenu. |
2. Favorito LA. Hémangiome vésical chez le patient atteint du syndrome de Klippel-Trenaunay-Weber. J Urol. 2003;29(2):149-50.
4. Klippel M, Trénaunay P. du naevus variqueux osteohypertrophique. Arch Gen Med (Paris)., 1900;185:641-72.
5. Weber FP. Hypertrophie hémangiectatique des membres: phlébartériectasie congénitale et varices dites congénitales. Br J Enfant Dis. 1918;15:13-7.
9. Mon JG, Schwartz RA, Janniger CK. Klippel-Trenaunay-Weber syndrome. Cutis. 1997;60(3):127-32. PMID: 9314616
1. Universidade Federal de Uberlândia, Uberlândia, MG, Brésil.
l’Établissement: Universidade Federal de Uberlândia, Uberlândia, MG, Brésil.