contexte
Le syndrome D’Alice au pays des merveilles (AIWS) est un trouble neurologique rare caractérisé par des distorsions de la perception visuelle (métamorphopsies), de l’image corporelle et de l’expérience du temps. Comme L’a noté dès 1955 John Todd, ces symptômes peuvent s’accompagner d’une déréalisation et d’une dépersonnalisation (1)., Les Patients souffrant D’AIWS peuvent finir par consulter un neurologue ou un psychiatre, bien que dans les deux spécialités, il ne soit pas aussi connu qu’il le mérite. Cela est dû au moins en partie au fait que les grandes classifications telles que le Manuel diagnostique et statistique des troubles mentaux ne le classent pas comme une catégorie diagnostique, tandis que d’autres, telles que la Classification internationale des maladies, n’y accordent qu’une attention limitée. Une autre raison peut être le nombre relativement faible de cas publiés., Une revue de la littérature existante, publiée en 2016, a indiqué que seulement 169 cas d’AIWS avaient été décrits depuis la conceptualisation du syndrome en 1955, ce qui se résume à un nombre moyen de 1,1 cas par an (4). Bien que ce nombre ait augmenté régulièrement au cours des dernières années, il se situe actuellement autour de 180. Il est à noter que ce sont tous des cas nécessitant des soins médicaux, c’est-à-dire ce que nous appelons des « cas cliniques ». »Pendant ce temps, des études de population à grande échelle indiquent que les symptômes de L’AISS sont ressentis assez fréquemment dans la population générale, avec des taux de prévalence au cours de la vie allant jusqu’à 30% (5-7)., Étant donné que ces derniers cas sont généralement de nature éphémère et transitoire, et impliquent rarement un comportement de recherche d « aide, ils sont appelés » cas non cliniques.” Des cas cliniques, environ 85% montrent l’implication d’une seule modalité sensorielle. Parmi eux, la majorité sont limités à un seul symptôme (par exemple, seulement micropsie ou seulement macrosomatognosie, etc.) (8). AIWS a de nombreuses étiologies différentes. Dans les cas cliniques, l’encéphalite (notamment due à une infection par le virus D’Epstein-Barr) est la cause la plus fréquente chez l’enfant; chez l’adulte, c’est la migraine (4)., Bien que le pronostic et les résultats dépendent en grande partie du trouble sous-jacent et de son aptitude au traitement, le fardeau causé par L’AIWS peut être important, même lorsque sa cause est assez inoffensive, comme dans le contexte de la fièvre ou de la consommation sporadique de cannabis. Cela est particulièrement vrai lorsque les symptômes perturbent gravement la perception sensorielle, interfèrent avec le fonctionnement quotidien et / ou évoquent des pensées catastrophiques (par exemple, que le monde a réellement changé d’une manière incompréhensible ou que les symptômes peuvent être indicatifs de démence ou de schizophrénie)., Étant donné que les cas non cliniques sont par définition Auto-limitatifs, et même dans 50% de tous les cas cliniques, l’assurance semble suffire, L’AIWS est parfois qualifiée de relativement inoffensive par nature (9). Cependant, étant donné que la recherche dans ce domaine en est encore à ses balbutiements et que L’AIWS pourrait bien être gravement sous-déclarée, il est trop tôt pour tirer des conclusions concernant son histoire naturelle et ses résultats « typiques”. De plus, nous savons déjà que L’AIWS peut être une manifestation de conditions graves et même mortelles, telles qu’un infarctus cérébral ou une tumeur cérébrale (10, 11)., Nous présentons ici un cas de Sai causé par la maladie sporadique de Creutzfeldt-Jakob (MCJ). Avec elle, nous ajoutons à la littérature en plein essor sur AIWS, et cherchons à contrer toute conclusion prématurée que ce syndrome peut a priori être caractérisé comme relativement « bénin” dans la nature.
Méthodes
Le patient nous décrivons était sous traitement dans notre propre pratique clinique. Étant donné qu’il est décédé dans les 2 mois suivant le début de ses symptômes de L’AIWS, nous avons obtenu le consentement écrit de sa famille pour publier.,
résultats–rapport de cas
en mars 2018, un homme de 68 ans s’est présenté à notre clinique externe de neurologie avec des symptômes visuels. Il avait des antécédents de chirurgie de la cataracte en 2004 et de surdité de perception à partir de 2006. Quelques semaines avant de le voir, il avait été référé à l’ophtalmologiste en raison d’une mauvaise perception de la profondeur et de la taille et d’une sensation de tension autour de la tête qui avait persisté pendant 1 mois. Il n’avait pas d’autres symptômes (neurologiques). L’évaluation ophtalmologique a indiqué un glaucome à angle ouvert avec des pressions intraoculaires de 39/27 mmHg ODS (référence 12-21 mmHg)., Son acuité visuelle était presque normale (DO 0·8 dpt, OS 0·9 dpt), bien que l’examen du champ visuel ait révélé plusieurs défauts légers (DO) qui ne correspondaient pas à ses symptômes. Sa pression oculaire s’est normalisée avec l’utilisation de bimatoprost et de gouttes oculaires dorzolamide/timolol. Cependant, au cours des semaines suivantes, les symptômes visuels sont devenus plus prononcés et particuliers. Lors de son renvoi à notre clinique externe, il a signalé que les objets en mouvement ralentissaient et accéléraient « comme dans un film de Charlie Chaplin » (connu sous le nom de durée prolongée et de phénomène de mouvement rapide, respectivement, c’est-à-dire,, variantes du groupe de distorsions temporelles). Parfois, il ne voyait même aucun mouvement (akinetopsia). De plus, il percevait des couleurs exceptionnellement vives (hyperchromatopsie) et changeant aléatoirement, comme un T-shirt rouge virant au vert « comme des lumières de fête foraine” (chloropsie/dyschromatopsie). Il a également vu des objets rétrécir ou gonfler jusqu’à une taille non naturelle, « comme s’il regardait dans un miroir funhouse” (micropsie et macropsie, zoom vision)., En plus de ces types de métamorphopsie, il a développé des paresthésies dans les mains et les pieds, une instabilité de la démarche et des dysfonctionnements de la concentration et de la mémoire. Il avait également de la difficulté à jouer de la flûte jazz et du saxophone, ce que nous considérions comme un signe inquiétant, étant donné qu’il était un musicien de jazz professionnel. Son discours est devenu moins fluide parce qu’il ne trouvait pas les bons mots ou ne les enchaînait pas pour faire des phrases, et son partenaire a remarqué des changements de comportement sous forme d’agitation et de labilité émotionnelle.,
bien que l’examen neurologique, les analyses de sang de routine et une IRM de la tête (y compris l’imagerie pondérée par diffusion, DWI) n’aient montré aucune anomalie, notre patient est devenu de plus en plus oublieux et désorienté, et a ensuite été admis à notre hôpital. Là, nous avons vu un homme auparavant éloquent qui souffrait maintenant d’aphasie, d’ataxie, de secousses myocloniques et d’une démence rapidement progressive., Une seconde IRM de tête a montré de faibles valeurs de coefficient de diffusion apparente (ADC) et une augmentation de la signalisation DWI dans le cortex pariéto-occipital gauche, caractéristique d’un signe de ruban cortical (Figure 1), tandis que l’électroencéphalogramme a montré des complexes d’ondes pointues périodiques (PSWC) sur la même zone (Figure 2). L’IRM avec présence de protéines 14-3-3 et de protéine tau élevée dans le liquide céphalo-rachidien a confirmé le diagnostic de la maladie de Creutzfeldt-Jakob (MCJ) sporadique probable, notamment la variante de Heidenhain., Nous avons exclu d’autres causes possibles, y compris les troubles auto-immuns, les infections, les tumeurs malignes, les encéphalopathies toxiques et métaboliques, et d’autres maladies neurodégénératives (notamment l’atrophie corticale postérieure et la démence à corps de Lewy). Nous n’avons pas effectué de tests génétiques parce que les antécédents familiaux étaient négatifs pour la maladie neurodégénérative, la MCJ en particulier. Malheureusement, il n’y avait pas d’options thérapeutiques. Conformément à ses souhaits, notre patient a été renvoyé chez lui, où il est décédé dans les 2 mois suivant le début de ses symptômes visuels. Nous n’avons pas pu obtenir le consentement de l’autopsie.,
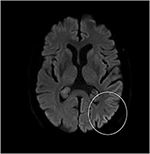
la Figure 1. Intensité élevée du signal du cortex pariéto-occipital gauche lors d’une IRM pondérée par diffusion (DWI; b = 1 000).
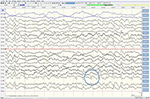
la Figure 2. EEG montrant des complexes d’ondes pointues périodiques sur la région pariéto-occipitale gauche, suggérant la MCJ.,
Discussion
ce qui rend ce cas unique, c’est que notre patient a présenté un nombre relativement important de métamorphopsies (qui, dans la littérature existante, n’est rapporté que dans 15% de tous les cas cliniques de SIA) et que la MCJ était la cause la plus probable. Étant donné qu’un diagnostic de MCJ définitive ne peut être établi post mortem que par autopsie, il était regrettable que nous n’ayons pas pu obtenir le consentement de l’autopsie. Néanmoins, les résultats de L’IRM se sont révélés positifs dans 83% des cas de MCJ probable (comparativement à 98% des cas de MCJ certaine) (12)., Par conséquent, même si L’IRM de la tête n’est pas l’étalon-or, un diagnostic de MCJ était toujours très probable chez notre patient, en particulier compte tenu de notre examen neurologique supplémentaire, qui n’a pas permis d’obtenir un autre diagnostic pouvant expliquer le tableau clinique, y compris son déclin rapide et ses résultats dramatiques.,
syndrome D’Alice au pays des merveilles
tel que décrit par John Todd en 1955, L’AIWS se caractérise par des distorsions perceptuelles rappelant les distorsions visuelles, les distorsions temporelles, les changements corporels, la déréalisation et la dépersonnalisation vécus par Alice pendant son séjour au pays des merveilles, comme décrit dans le livre pour Enfants Classique, Todd, et avant lui, Lipmann (14), ont suggéré que Carroll – de son vrai nom Charles Lutwidge Dodgson-avait trouvé l’inspiration pour ces phénomènes particuliers dans les distorsions perceptuelles qu’il avait lui-même expérimentées dans le contexte de la migraine. Bien que cette hypothèse ait été suivie par plusieurs traités historiques alléguant que la migraine était effectivement la source la plus probable des expériences de Dodgson (15, 16), d’autres ont avancé l’hypothèse tout aussi alléchante que, dans son cas, c’était plutôt l’épilepsie qui les causait (17) ou peut-être la toxicomanie (18)., Bien que nous ne saurions probablement jamais avec certitude ce qui a poussé Dodgson à ressentir ces symptômes, une reconstruction minutieuse de ses antécédents médicaux indique que leur source la plus probable était une maladie infectieuse, dont l’auteur a souffert à de nombreuses reprises depuis l’enfance, littéralement jusqu’au dernier jour de sa vie (19)., Il est très probable que Dodgson avait lui-même expérimenté ces distorsions (plutôt que d’avoir basé les expériences de son héroïne sur des informations de troisième main), avec un énorme 13 symptômes différents décrits tout au long du livre, bien avant que Todd n’ait inventé le concept D’AIWS.,
pour comprendre ce qui distingue les AIW des autres syndromes et troubles perceptifs, nous devons réaliser que les distorsions diffèrent des hallucinations et des illusions en ce qu’elles ne sont ni des percepts nouvellement formés de quelque chose qui n’est pas là (hallucination), ni des objets ou des scènes réels jugés à tort comme autre chose (illusion). Au lieu de cela, les distorsions sont des percepts, vécus par un individu éveillé, qui sont basés sur des stimuli appropriés du monde extérieur, dont un aspect très spécifique est modifié de manière cohérente ., Aujourd’hui, on connaît quelque 55 types différents de distorsion perceptuelle qui peuvent se produire dans le contexte de L’AIWS (4). Les plus fréquemment signalés sont la micropsie (voir les choses plus petites qu’elles ne le sont), la macropsie (voir les choses plus grandes qu’elles ne le sont), la téléopsie (voir les choses plus loin qu’elles ne le sont) et la dysmorphopsie (voir les lignes droites comme ondulées). La disposition selon laquelle ces aspects sont modifiés de manière cohérente signifie que la distorsion s’applique à tout ce qui se trouve dans la portée visuelle du patient., Ainsi, les personnes souffrant de plagiopsie verront généralement toutes les choses comme inclinées, dans la même direction et sous le même degré, tandis que celles atteintes de prosopométamorphopsie verront généralement une altération particulière dans tous les visages qu’elles perçoivent, tels que des sourcils lourds ou un œil nettement enlevé au nez ou des visages humains se transformant en visages de dragon (21, 22). La nature de ces distorsions peut varier au fil du temps, mais une fois présentes, toutes les choses perçues semblent déformées de la même manière.

le Tableau 1., Définitions de l’hallucination, de l’illusion et de la distorsion .
La distinction entre l’hallucination, illusion et de la distorsion n’est pas seulement une question théorique. Au contraire, on pense que leurs différences phénoménologiques reflètent des différences dans les mécanismes sous-jacents. Physiopathologiquement, les distorsions perceptuelles sont attribuées à des lésions structurelles ou fonctionnelles de parties discrètes du réseau perceptuel, telles que l’aire V4 chez l’hyperchromatopsie et V5 chez l’akinétopsie (23)., Il existe huit groupes connus d’étiologie sous-jacente, comprenant les maladies infectieuses (encéphalite), les lésions du système nerveux central (SNC) (accident vasculaire cérébral, tumeur cérébrale), les lésions du système nerveux périphérique (SNP) (maladie oculaire, maladie de l’oreille moyenne), les troubles neurologiques paroxystiques (épilepsie, migraine), les troubles psychiatriques (dépression, schizophrénie), les médicaments, les substances illicites et un groupe divers qui comprend l’hypnagogie et la privation sensorielle (4)., Ainsi, les soi-disant « cas cliniques” d’AIWS justifient toujours un examen diagnostique, y compris une consultation neurologique et psychiatrique, une IRM de la tête et une EEG et, le cas échéant, d’autres examens auxiliaires (par exemple, examen ophtalmologique, consultation ORL). Bien que des traitements fondés sur des preuves ne soient pas encore développés, il est de bonne pratique clinique de cibler la thérapie sur la cause sous-jacente (probable) (4, 19).
maladie de Creutzfeldt-Jakob
à notre connaissance, L’AIWS n’a jamais été signalée dans le contexte de la MCJ. Avec 1-1.,5 cas par million d’habitants par année, la MCJ est une maladie extrêmement rare qui est mortelle en un an chez environ 90% des personnes touchées (24). L’incidence maximale se situe dans la septième décennie, alors que les cas plus jeunes (20-40 ans) ou plus âgés (>80 ans) sont beaucoup moins fréquents (25). La MCJ fait partie des encéphalopathies spongiformes transmissibles ou des maladies à prions. Les maladies à prions sont caractérisées par le dépôt d’une isoforme anormalement mal repliée de la protéine prion native, codée par le gène prion (PRNP) sur le chromosome 20 humain., Le mécanisme de déclenchement de ce changement conformationnel est inconnu, mais l’accumulation de cette protéine prion anormale entraîne une dégénérescence neuronale, une gliose astrocytaire et des changements spongiformes, accompagnés d’un déclin cognitif rapide et se terminant par la mort.,la MCJ, qui représente environ 85% des cas; (ii) la MCJ génétique, qui représente 10 à 15% des cas et est associée à des mutations du gène prion (PRNP); (iii) la forme iatrogène de la MCJ, qui représente environ 1% des cas et est le plus souvent associée à un traitement antérieur par des hormones d’origine hypophysaire humaine ou des greffes de la consommation de produits de boeuf contaminés par l’agent causal de l’encéphalopathie spongiforme bovine (ESB) ou « maladie de la vache folle., »De plus, sur la base de la présentation clinique initiale, six phénotypes ont été identifiés, appelés cognitifs, visuels (variante Heidenhain), affectifs, classiques, atactiques (variante Oppenheimer-Brownell) et indéterminés (26). La variante Heidenhain a été décrite pour la première fois dans les années 1920 et nommée d’après le physiologiste et histologiste allemand Rudolf Heidenhain (1834-1897)., Représentant environ 20% de tous les cas de MCJ sporadique (26-28), elle se caractérise par des symptômes visuels isolés au début de la maladie, tels que des anomalies du champ visuel et une perte d’acuité visuelle, qui sont considérés comme des reflets d’une atteinte précoce du cortex occipital (29). Étant donné que ces défauts visuels peuvent précéder les symptômes cognitifs et moteurs de plusieurs semaines, il n’est pas rare que la MCJ ne soit même pas suspectée dans de tels cas et que les patients soient initialement dirigés vers un ophtalmologiste.,
Conclusion
Nous concluons que la possibilité d’une variante Heidenhain de la MCJ, bien qu’extrêmement rare, doit être abordée dans le diagnostic différentiel de L’AIWS, en particulier en présence d’un déclin cognitif rapide. De plus, nous concluons que les SSAI peuvent être relativement inoffensifs dans la majorité des cas, mais que les cas « cliniques” justifient toujours un diagnostic approprié avant que cette conclusion puisse être tirée.
Limitations
notre connaissance des nombreuses causes et manifestations de L’AIWS en est encore à ses balbutiements., Le syndrome a été conceptualisé pour la première fois en 1955, mais seules les dernières années ont vu une augmentation constante du nombre de publications sur ce groupe disparate de phénomènes. Étant donné que la région est encore en pleine mutation, nos réflexions théoriques sur L’AIWS, bien qu’à l’état de l’art, doivent être considérées comme préliminaires. Deuxièmement, le fait que nous présentons ici le premier rapport de cas sur L’AIWS dans le contexte de la MCJ ne signifie pas nécessairement que notre patient a été la première personne à souffrir de ces deux maladies rares., Notamment, on peut s’attendre à ce que la variante Heidenhain de la MCJ se présente plus fréquemment avec les métamorphopsies. Par conséquent, notre incapacité à trouver des descriptions de cas antérieurs de symptômes de L’AISS dans le contexte de la MCJ pourrait bien être due à une sous-déclaration plutôt qu’à une rareté réelle de la combinaison. Une troisième et dernière limite est que nous n’avons pas pu obtenir le consentement de l’autopsie, ce qui aurait été nécessaire pour établir un diagnostic de MCJ.
énoncé D’éthique
le consentement écrit à la publication a été obtenu de la famille du patient.,
Contribution à L’énoncé de terrain
Le syndrome D’Alice au pays des merveilles (AIWS) est un trouble neurologique rare caractérisé par des distorsions de la perception visuelle, de l’image corporelle et de l’expérience du temps. Les gens peuvent voir des choses plus petites qu’elles ne le sont, sentir que leur corps change de taille ou ressentir l’un des nombreux autres symptômes du syndrome. Comme il existe également de nombreuses causes connues d’AISS, le diagnostic nécessite un examen neurologique approfondi. Chez les enfants, la cause la plus fréquente est l’inflammation du cerveau; chez les adultes, c’est la migraine., Dans la littérature médicale, pas plus de 180 patients individuels ont été décrits, dont 50% se sont rétablis. Cependant, des études de population à grande échelle indiquent que jusqu’à 30% de tous les individus en bonne santé présentent des symptômes éphémères et transitoires de L’AISS. En conséquence, AIWS est généralement considéré comme relativement inoffensif dans la nature. Nous présentons ici la première description de cas d’un homme âgé qui souffrait D’une SIA due à la maladie de Creutzfeldt-Jakob (MCJ), une maladie neurodégénérative extrêmement rare., Notre patient a rapidement développé un État de démence et est décédé dans les 2 mois suivant le début de ses symptômes visuels. Nous concluons que L’AIWS n’est pas toujours inoffensive et que la MCJ, bien qu’extrêmement rare, doit être dans l’esprit des médecins lorsque les symptômes visuels typiques de L’AIWS s’accompagnent d’un déclin cognitif rapide.,
contributions des auteurs
TN a contribué à la conception et à la conception de l’œuvre, ainsi qu’à l’acquisition, à l’analyse et à l’interprétation des données pour l’œuvre, a rédigé et révisé l’œuvre, a donné son approbation finale pour la publication de la version finale et a accepté d’être responsable de tous les aspects de l’œuvre en veillant à ce que les questions liées à l’exactitude ou à l’intégrité de toute partie de l’œuvre fassent l’objet d’une enquête et d’une résolution appropriées., BtM et SvdW ont contribué à la conception et à la conception du travail, ainsi qu’à l’acquisition, à l’analyse et à l’interprétation des données pour le travail, ont révisé le travail, ont donné l’approbation finale pour la publication de la version finale et ont convenu d’être responsables de tous les aspects du travail en veillant à ce que les questions liées à l’exactitude ou à l’intégrité de toute partie du travail fassent l’objet d’une enquête et d’une résolution appropriées., JDB contribué à la conception et à la conception de l’ouvrage, et l’analyse et l’interprétation des données pour le travail, rédigé et révisé le travail, a donné l’approbation finale de la version finale qui sera publiée, et accepté d’être responsable de tous les aspects du travail en veillant à ce que les questions liées à l’exactitude ou l’intégrité de toute partie des travaux qui sont bien étudiés et résolus.,
déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière pouvant être interprétée comme un conflit d’intérêts potentiel.
1. Todd J. Le syndrome D’Alice au pays des merveilles. Peut-Med Assoc J. (1955) 73:701-4.
PubMed Abstract | Google Scholar
2. Association Américaine De Psychiatrie. Manuel diagnostique et statistique des troubles mentaux. 5e ed., Washington, DC: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
CrossRef Full Text | Google Scholar
3. World Health Organization. The International Classification of Diseases, 10th Revision. Geneva: World Health Organization (1992).
Google Scholar
5. Abe K, Suzuki T., Prévalence de certains symptômes à l’adolescence et à la maturité: phobies sociales, symptômes d’anxiété, illusions épisodiques et idée de référence. Psychopathologie. (1986) 19:200–5. doi: 10.1159/000284448
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
8. Lanska JR, LANSKA DJ. Alice au pays des merveilles syndrome: somesthésique vs perturbation perceptuelle visuelle. Neurologie. (2013) 80:1262–4. doi: 10.1212 / WNL.,0b013e31828970ae
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
11. Bender MB, Kanzer MG. Métamorphopsie et autres troubles psychovisuels chez un patient atteint d’une tumeur du cerveau. Arch Neurol De La Psychiatrie. (1941) 45:481–5. doi: 10.1001 / archneurpsyc.1941.02280150089005
CrossRef Texte Intégral | Google Scholar
13. Les Aventures de Carroll L. Alice au pays des merveilles. Londres: MacMillan (1865).,
Google Scholar
16. Podoll K, Robinson D. Migraine Art. The Migraine Experience From Within. Berkeley, CA: North Atlantic Books (2008).
Google Scholar
18. Carmichael C. Wonderland revisited. London Miscellany. (1996) 28:19–28.
Google Scholar
19. Blom JD. Alice in Wonderland Syndrome., Heidelberg: Springer (2019).
Google Scholar
22. Funatsu N, Hayakawa M, Tokuda N, Toyoda K. prosopométamorphopsie transitoire limitée à l’œil gauche causée par une ischémie au splénium droit du corps calleux. Intern Med. (2017) 56:2933–5. doi: 10.2169 / internalmedicine.8295-16
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
le 24., Appleby BS, Appleby KK, Crain BJ, ONYIKE CU, Wallin MT, Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol. (2009) 66:208-15. doi: 10.1001/archneurol.2008.533
CrossRef Full Text | Google Scholar
26. Heidenhain A. études cliniques et anatomiques sur une maladie organique particulière du système nerveux central praesenium. Z GE Neurol Psychiatre. (1929) 118: 49-114.doi: 10.,1007/BF02892896
CrossRef Full Text | Google Scholar
28. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, et al. Revisiting the Heidenhain variant of Creutzfeldt-Jakob disease: evidence for prion type variabiliy influencing clinical course and laboratory findings. J Alzheimers Dis. (2016) 50:465–76. doi: 10.3233/JAD-150668
CrossRef Full Text | Google Scholar
29., Cooper SA, Murray KL, Heath CA, Va RG, Chevalier de la RSG. Symptômes visuels isolés au début de la maladie sporadique de Creutzfeldt-Jakob: le phénotype clinique de la » variante de Heidenhain. »Br J Ophthalmol. (2005) 89:1341–2. doi: 10.1136 / bjo.2005.074856
PubMed Abstract | CrossRef Texte Intégral | Google Scholar