Spate
sindromul Alice în țara Minunilor (AIWS) este o tulburare neurologică rară caracterizată prin distorsiuni de percepție vizuală (metamorphopsias), imaginea corporală, și experiența de timp. După cum sa menționat încă din 1955 de John Todd, aceste simptome pot fi însoțite de derealizare și depersonalizare (1)., Pacienții care suferă de AIWS pot ajunge să consulte un neurolog sau un psihiatru, deși în ambele specialități nu este la fel de cunoscut pe cât merită să fie. Aceasta este, cel puțin parțial, datorită faptului că principalele clasificări, cum ar fi Manualul de Diagnostic și Statistică a Tulburărilor Mentale nu lista ca o categorie diagnostică, în timp ce altele, cum ar fi Clasificarea Internațională a Bolilor plătească limitat doar o atenție la ea. Un alt motiv poate fi numărul relativ mic de cazuri publicate., O revizuire a literaturii existente, publicate în 2016, a indicat că doar 169 de cazuri de AIWS a fost descris de sindromul lui conceptualizare în 1955, care se reduce la un număr de 1,1 cazuri pe an (4). Deși acest număr a crescut constant în ultimii ani, în prezent se află în jur de 180. De remarcat, toate acestea sunt cazuri care necesită îngrijiri medicale, adică ceea ce numim „cazuri clinice.”Între timp, studiile la scară largă ale populației indică faptul că simptomele AIWS sunt experimentate destul de frecvent în populația generală, cu rate de prevalență pe viață de până la 30% (5-7)., Deoarece aceste din urmă cazuri sunt de obicei trecătoare și tranzitorii în natură, și rareori implică un comportament care caută ajutor, acestea sunt denumite „cazuri nonclinice.”Dintre cazurile clinice, aproximativ 85% arată implicarea unei singure modalități senzoriale. Dintre acestea, majoritatea sunt limitate la un singur simptom (de exemplu, numai micropsia sau numai macrosomatognosia etc.) (8). AIWS are multe etiologii diferite. În cazurile clinice, encefalita (în special din cauza infecției cu virusul Epstein-Barr) este cea mai frecventă cauză la copii; la adulți, este migrenă (4)., Deși prognosticul și rezultatul depind în mare măsură de tulburarea de bază și de capacitatea sa de tratament, povara cauzată de AIWS poate fi substanțială, chiar și atunci când cauza sa este destul de inofensivă, ca în contextul febrei sau al consumului sporadic de canabis. Acest lucru este valabil mai ales atunci când simptomele perturbă grav percepția senzorială, interferează cu funcționarea zilnică și/sau evocă gânduri catastrofale (de exemplu, că lumea s-a schimbat într-un mod incomprehensibil sau că simptomele pot indica demență sau schizofrenie)., Deoarece cazurile non-clinice sunt, prin definiție, auto-limitate și chiar și în 50% din toate cazurile clinice, reasigurarea pare să fie suficientă, AIWS este uneori caracterizat ca fiind relativ inofensiv în natură (9). Cu toate acestea, de cercetare în acest domeniu este încă în fază incipientă, și AIWS poate fi grav subestimate, este prea devreme pentru a trage concluzii cu privire la istoria naturală și „tipic” rezultatul. Mai mult, știm deja că AIWS poate fi o manifestare a unor afecțiuni severe și chiar letale, cum ar fi infarctul cerebral sau tumora cerebrală (10, 11)., Prezentăm aici un caz de AIWS cauzat de boala sporadică Creutzfeldt-Jakob (CJD). Cu aceasta, adăugăm la literatura înfloritoare despre AIWS și căutăm să contracarăm orice concluzii premature că acest sindrom poate fi a priori caracterizat ca fiind relativ „benign” în natură.
metode
pacientul pe care îl descriem a fost sub tratament în propria noastră practică clinică. Din moment ce a murit în termen de 2 luni de la simptomele sale de AIWS au început, am obținut acordul scris de a publica de la familia sa.,în martie 2018, un bărbat în vârstă de 68 de ani a raportat la clinica noastră de neurologie cu simptome vizuale. El a avut o istorie de chirurgie cataractei în 2004 și percepția surditate începând din 2006. Cu câteva săptămâni înainte de a-l vedea, a fost trimis la oftalmolog din cauza percepției slabe a adâncimii și dimensiunii și a unei senzații strânse în jurul capului care a persistat timp de 1 lună. Nu avea alte simptome (neurologice). Evaluarea oftalmologică a indicat un glaucom cu unghi deschis cu presiuni intraoculare de 39/27 mmHg ODS (referință 12-21 mmHg)., Acuitatea sa vizuală a fost aproape normală (OD 0 * 8 dpt, OS 0 * 9 dpt), deși examinarea câmpului vizual a evidențiat mai multe defecte ușoare (OD) care nu erau în concordanță cu simptomele sale. Presiunea oculară sa normalizat cu utilizarea picăturilor de ochi bimatoprost și dorzolamidă / timolol. Cu toate acestea, pe parcursul următoarelor săptămâni, simptomele vizuale au devenit mai pronunțate și mai ciudate. La trimiterea la clinica noastră de ambulatoriu, el a raportat că obiectele în mișcare încetinesc și accelerează „ca într-un film Charlie Chaplin” (cunoscut sub numele de durată prelungită și respectiv fenomen de mișcare rapidă, adică.,, variante ale grupului de distorsiuni de timp). Uneori chiar nu a reușit să vadă nici o mișcare (akinetopsia). În plus, el culori percepute ca extrem de luminoase (hyperchromatopsia) și schimbă aleatoriu, cum ar fi un tricou roșu de cotitură verde „ca bâlci lumini” (chloropsia/discromatopsie). De asemenea, a văzut obiecte care se micșorează sau se umflă până la o dimensiune nenaturală, „ca și cum ar privi într-o oglindă funhouse” (micropsia și macropsia, zoom vision)., În plus față de aceste tipuri de metamorfopsie, el a dezvoltat parestezii în mâini și picioare, instabilitate de mers și tulburări de concentrare și memorie. De asemenea, a avut dificultăți în a cânta la flaut și saxofon de jazz, pe care le-am considerat un semn de rău augur, având în vedere că era un muzician profesionist de jazz. Discursul său a devenit mai puțin fluent, deoarece nu a putut găsi cuvintele potrivite sau nu le-a strâns împreună pentru a face propoziții, iar partenerul său a observat schimbări comportamentale sub formă de agitație și labilitate emoțională.,deși examenul neurologic, testele de sânge de rutină și un RMN la cap (inclusiv imagistica ponderată prin difuzie, DWI) nu au arătat anomalii, pacientul nostru a devenit din ce în ce mai uitat și dezorientat și a fost ulterior internat în spitalul nostru. Acolo am văzut un om elocvent anterior, care acum suferea de afazie, ataxie, smucituri mioclonice și o demență rapid progresivă., Un al doilea cap RMN a arătat scăzut coeficientul de difuzie aparentă (ADC) a valorilor și a crescut DWI-semnalizare în stânga parieto-occipital cortex, caracteristice unei corticale panglică semn (Figura 1), în timp ce electroencefalograma arătat periodice ascuțite-val complexe (PSWC) în aceeași zonă (Figura 2). RMN-ul cu prezența proteinelor 14-3-3 și a proteinei tau crescute în lichidul cefalorahidian a susținut diagnosticul bolii sporadice probabile Creutzfeldt-Jakob (CJD), în special varianta Heidenhain., Am exclus alte cauze posibile, inclusiv tulburări auto-imune, infecții, afecțiuni maligne, encefalopatii toxice și metabolice și alte boli neurodegenerative (în special atrofia corticală posterioară și demența cu corpi Lewy). Nu am efectuat teste genetice, deoarece istoricul familial a fost negativ pentru boala neurodegenerativă, în special CJD. Din păcate, nu au existat opțiuni terapeutice. În conformitate cu dorințele sale, pacientul nostru a fost externat acasă, unde a murit în termen de 2 luni de la debutul simptomelor sale vizuale. Nu am putut obține consimțământul autopsiei.,
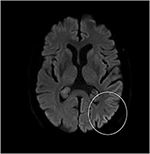
Figura 1. Intensitate ridicată a semnalului cortexului parieto-occipital stâng pe RMN ponderat cu difuzie (DWI; b = 1.000).
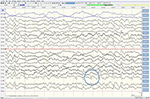
Figura 2. EEG care prezintă complexe periodice cu unde ascuțite peste regiunea parieto-occipitală stângă, sugestive pentru CJD.,
Discuții
ceea Ce face acest caz unic, este că pacientul s-a prezentat cu un număr relativ mare de metamorphopsias (care, în literatura existente, este raportat în numai 15% din toate cazurile clinice de AIWS) și că CJD a fost cauza cea mai probabilă. Deoarece un diagnostic de CJD definit poate fi stabilit doar post mortem prin autopsie, a fost regretabil că nu am putut obține consimțământul autopsiei. Cu toate acestea, rezultatele RMN s-au dovedit a fi pozitive în 83% din cazurile de CJD probabil (față de 98% din cazurile de CJD definit) (12)., Prin urmare, chiar dacă RMN-ul capului nu este standardul de aur, un diagnostic de CJD era încă foarte probabil la pacientul nostru, în special având în vedere munca noastră neurologică suplimentară, care nu a reușit să producă un diagnostic alternativ care ar putea explica tabloul clinic, inclusiv declinul rapid și rezultatul dramatic.,
Sindromul Alice în țara Minunilor
Cum este descris de John Todd, în 1955, AIWS se caracterizează prin distorsiuni perceptuale amintește de distorsiuni vizuale, distorsiuni temporale, modificări corporale, derealization, și depersonalizare experimentat de Alice în timpul șederii sale în țara Minunilor, după cum este descris în Lewis Carroll clasic de carte pentru copii Aventurile lui Alice în țara Minunilor (1, 13)., Todd, și înaintea lui, Lipmann (14), a sugerat că Carroll–al cărui nume real era Charles Lutwidge Dodgson–a găsit inspirație pentru aceste fenomene ciudate în distorsiunile perceptuale pe care le-a experimentat în contextul migrenei. Deși această ipoteză a fost urmat de mai multe istorice tratate susținând că migrena a fost într-adevăr cea mai probabilă sursă a lui Dodgson experiențe (15, 16), altele au prezentat la fel de tentantă ipoteza că, în cazul său, a fost destul de epilepsie pe care le-a cauzat (17) sau, poate, abuzul de substanțe (18)., Deși probabil nu vom sti niciodata sigur ce a făcut Dodgson prezentați aceste simptome, printr-o reconstrucție de istoricul său medical indică faptul că lor cel mai probabil sursa de boli infecțioase, din care autorul a suferit, în numeroase ocazii, începând încă din copilărie, literalmente, până în ultima zi a vieții sale (19)., Este foarte probabil ca Dodgson a experimentat aceste distorsiuni se (mai degrabă decât bazate pe eroina lui experiențele pe informații la mâna a treia), cu un enorm 13 diferite simptome descrise de-a lungul cărții, cu mult înainte de Todd a venit cu conceptul de AIWS.,
Pentru a înțelege ceea ce face AIWS stea afară din alte perceptuale sindroame și tulburări, trebuie să realizăm că denaturările diferă de halucinații și iluzii în care acestea nu sunt nici nou format de perceptie din ceva care nu există (halucinații), nici obiecte reale sau scene eronat considerate a fi altceva (iluzie). În schimb, distorsiunile sunt percepții, experimentate de un individ treaz, care se bazează pe stimuli adecvați din lumea exterioară, dintre care un aspect extrem de specific este modificat într-o manieră consecventă ., Astăzi, sunt cunoscute aproximativ 55 de tipuri diferite de distorsiuni perceptuale care pot apărea în contextul AIWS (4). Cele mai frecvent raportate sunt micropsie (a vedea lucruri mai mici decât sunt), macropsia (vede lucrurile mai mari decât sunt), teleopsia (vezi lucrurile mai departe decât sunt), și dysmorphopsia (vezi liniile drepte ca ondulate). Prevederea că astfel de aspecte sunt modificate în mod consecvent înseamnă că distorsiunea se aplică oricărui lucru din domeniul vizual al pacientului., Astfel, persoanele care suferă de plagiopsia va vedea de obicei toate lucrurile ca inclinat, în aceeași direcție, și în același grad, în timp ce cei cu prosopometamorphopsia va vedea de obicei o anumită modificare în toate chipurile ei percep, cum ar fi sprâncene sau un ochi care este semnificativ răpit la nas sau chipuri umane schimbare în dragon fețe (21, 22). Natura acestor distorsiuni poate varia în timp, dar odată prezente, toate lucrurile percepute apar ca și cum ar fi distorsionate într-un mod similar.

Tabelul 1., Definițiile halucinației, iluziei și distorsiunii .distincția dintre halucinație, iluzie și distorsiune nu este doar o problemă academică. Dimpotrivă, se consideră că diferențele lor fenomenologice reflectă diferențele dintre mecanismele care stau la baza acestora. Din perspectiva fiziopatologică, distorsiuni perceptuale sunt atribuite structurale, funcționale sau leziuni de părți distincte de percepție de rețea, cum ar fi zona V4 în hyperchromatopsia și V5 în akinetopsia (23)., Există opt grupuri de etiologia de fond al sistemului, care cuprinde boli infecțioase (encefalita), sistemul nervos central (SNC) leziuni (accident vascular cerebral, tumori cerebrale), sistemul nervos periferic (SNP) leziuni (boala de ochi, de mijloc-boala ureche), paroxistice tulburări neurologice (epilepsie, migrenă), tulburări psihice (depresie, schizofrenie), medicamente, substanțe ilicite, și diverse grup care include hypnagogia și privarea senzorială (4)., Astfel, așa-numitele „cazuri clinice” de AIWS întotdeauna mandat de diagnosticare, inclusiv neurologice și psihiatrice consultare, cap RMN si EEG, și, atunci când este indicat, alte auxiliare investigații (de exemplu, examenul oftalmologic, ORL consultare). Deși tratamentele bazate pe dovezi nu sunt încă dezvoltate, este o bună practică clinică să se urmărească terapia la cauza (probabilă) de bază (4, 19).
boala Creutzfeldt-Jakob
Din cunoștințele noastre, AIWS nu a fost raportat înainte în contextul CJD. Cu 1-1.,5 cazuri la un milion de locuitori pe an, CJD este o boală extrem de rară care este fatală în decurs de un an la aproximativ 90% dintre cei afectați (24). Incidența maximă este în deceniul al șaptelea, în timp ce cazurile mai tinere (20-40 ani) sau mai în vârstă (>80 ani) sunt mult mai puțin frecvente (25). CJD aparține encefalopatiilor spongiforme transmisibile sau bolilor prionice. Bolile prionice se caracterizează prin depunerea unei izoforme anormal de greșite a proteinei prionice native, care este codificată de gena prionică (PRNP) pe cromozomul uman 20., Mecanismul de declanșare a acestei schimbări conformaționale nu este cunoscut, dar acumularea acestei proteine prionice anormale duce la degenerare neuronală, glioză astrocitică și modificări spongiforme, însoțite de declin cognitiv rapid și care se termină cu moartea.,aproximativ 85% din cazuri; (ii) genetice CJD, care reprezintă 10-15% din cazuri și este asociat cu mutații ale genei prionice (PRNP); (iii) iatrogene formă de CJD, care reprezintă aproximativ 1% din cazuri și este cel mai frecvent asociat cu un tratament anterior cu pituitare umane derivate hormoni sau uman dura-mater grefe; și (iv) varianta CJD (vCJD), care este un roman umane bolile prionice găsite aproape exclusiv în marea BRITANIE, și a fost legată de consumul de carne de vită produse contaminate cu agentul cauzator de encefalopatie spongiformă bovină (ESB) sau „boala vacii nebune.,”Mai mult, pe baza prezentării clinice inițiale, au fost identificate șase fenotipuri, numite cognitive, vizuale (varianta Heidenhain), afective, clasice, atactice (varianta Oppenheimer-Brownell) și nedeterminate (26). Varianta Heidenhain a fost descrisă pentru prima dată în anii 1920 și numită după fiziologul și histologul German Rudolf Heidenhain (1834-1897)., Reprezentând aproximativ 20% din toate cazurile de CJD sporadice (26-28), se caracterizează prin simptome vizuale izolate la debutul bolii, cum ar fi defectele câmpului vizual și pierderea acuității vizuale, care sunt considerate a fi reflecții ale implicării precoce a cortexului occipital (29). Deoarece aceste defecte vizuale pot preceda simptomele cognitive și motorii cu câteva săptămâni, nu este neobișnuit ca CJD să nu fie nici măcar suspectat în astfel de cazuri și că pacienții sunt inițial referiți la un oftalmolog.,concluzionăm că posibilitatea unei variante Heidenhain a CJD, deși extrem de rară, trebuie să fie distrați în diagnosticul diferențial al AIWS, în special în prezența declinului cognitiv rapid. Mai mult, concluzionăm că AIWS poate fi relativ inofensiv în majoritatea cazurilor, dar că cazurile „clinice” justifică întotdeauna o analiză corectă a diagnosticului înainte de a putea fi trasă această concluzie.
limitări
cunoștințele noastre despre numeroasele cauze și manifestări ale AIWS sunt încă în fază incipientă., Sindromul a fost conceptualizat pentru prima dată în 1955, dar numai în ultimii ani au cunoscut o creștere constantă a numărului de publicații despre acest grup disparat de fenomene. Deoarece zona este încă foarte mult în flux, reflecțiile noastre teoretice asupra AIWS, deși de ultimă oră, trebuie considerate preliminare în natură. În al doilea rând, faptul că prezentăm primul raport de caz pe AIWS în contextul CJD nu implică neapărat că pacientul nostru a fost prima persoană care suferă de aceste două afecțiuni rare., În special, varianta Heidenhain a CJD poate fi de așteptat să se prezinte mai frecvent cu metamorfopsii. Prin urmare, incapacitatea noastră de a găsi descrieri anterioare ale simptomelor AIWS în contextul CJD se poate datora mai degrabă subraportării decât unei rarități reale a combinației. O a treia și ultima limitare este că nu am putut obține consimțământul autopsiei, ceea ce ar fi fost necesar pentru a face un diagnostic de CJD definit.
declarație etică
consimțământul scris de publicare a fost obținut din partea familiei pacientului.,sindromul Alice in Tara Minunilor (AIWS) este o tulburare neurologică rară caracterizată prin distorsiuni ale percepției vizuale, a imaginii corpului și a experienței timpului. Oamenii pot vedea lucruri mai mici decât sunt, simt corpul lor modifica în dimensiune sau experiență oricare dintre sindromul numeroase alte simptome. Deoarece există, de asemenea, multe cauze cunoscute ale AIWS, diagnosticul necesită o analiză neurologică amănunțită. La copii, cea mai frecventă cauză este inflamația creierului, la adulți, este migrenă., În literatura medicală nu au fost descrise mai mult de 180 de pacienți individuali, dintre care 50% s-au recuperat. Cu toate acestea, studiile la scară largă ale populației indică faptul că simptomele trecătoare și tranzitorii ale AIWS sunt experimentate de până la 30% din toți indivizii sănătoși. În consecință, AIWS este în general considerat destul de inofensiv în natură. Prezentăm aici prima descriere a unui bărbat în vârstă care a suferit de AIWS din cauza bolii Creutzfeldt-Jakob (CJD), o tulburare neurodegenerativă extrem de rară., Pacientul nostru a dezvoltat rapid o stare de demență, și a murit în termen de 2 luni de la simptomele sale vizuale au început. Concluzionăm că AIWS nu este întotdeauna inofensiv și că CJD, deși extrem de rar, trebuie să fie în mintea medicilor atunci când simptomele vizuale tipice pentru AIWS sunt însoțite de un declin cognitiv rapid.,TN a contribuit la conceperea și proiectarea lucrării și la achiziționarea, analiza și interpretarea datelor pentru lucrare, a elaborat și revizuit lucrarea, a dat aprobarea finală pentru versiunea finală care urmează să fie publicată și a fost de acord să fie responsabil pentru toate aspectele lucrării, asigurându-se că întrebările legate de acuratețea sau integritatea oricărei părți a lucrării sunt investigate și rezolvate în mod corespunzător., BtM, și SvdW contribuit la conceperea și proiectarea de muncă, precum și pentru achiziția, analiza și interpretarea de date pentru munca, revizuit de lucru, a dat aprobarea finală pentru versiunea finală să fie publicate, și a fost de acord să fie responsabil pentru toate aspectele legate de munca în a se asigura că întrebările referitoare la acuratețea sau integritatea orice parte a lucrării sunt corect investigate și rezolvate., JDB a contribuit la conceperea și proiectarea de muncă, și de a analiza și interpretarea de date pentru muncă, elaborat și revizuit de lucru, a dat aprobarea finală pentru versiunea finală să fie publicate, și a fost de acord să fie responsabil pentru toate aspectele legate de munca în a se asigura că întrebările referitoare la acuratețea sau integritatea orice parte a lucrării sunt corect investigate și rezolvate.,
Declarație privind conflictul de interese
autorii declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
1. Todd J. sindromul lui Alice în țara Minunilor. Poate Med Assoc J. (1955) 73: 701-4.
PubMed Abstract | Google Scholar
2. Asociația Americană De Psihiatrie. Manual de Diagnostic și Statistic al tulburărilor mintale. A 5-a ed., Washington, DC: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
CrossRef Full Text | Google Scholar
3. World Health Organization. The International Classification of Diseases, 10th Revision. Geneva: World Health Organization (1992).
Google Scholar
5. Abe K, Suzuki T., Prevalența unor simptome în adolescență și maturitate: fobii sociale, simptome de anxietate, iluzii episodice și idee de referință. Psihopatologie. (1986) 19:200–5. doi: 10.1159/000284448
PubMed Abstract | CrossRef Textul Complet | Google Scholar
8. Lanska JR, Lanska DJ. Sindromul Alice în țara Minunilor: tulburări perceptuale somestetice vs vizuale. Neurologie. (2013) 80:1262–4. doi: 10.1212 / WNL.,0b013e31828970ae
PubMed Abstract | CrossRef Textul Complet | Google Scholar
11. Bender MB, KANZER MG. Metamorfopsia și alte tulburări psihovisuale la un pacient cu tumoare a creierului. Arch Neurol Psychiatry. (1941) 45:481–5. doi: 10.1001/archneurpsyc.1941.02280150089005
CrossRef Textul Complet | Google Scholar
13. Aventurile lui Carroll L. Alice în țara Minunilor. Londra: MacMillan (1865).,
Google Scholar
16. Podoll K, Robinson D. Migraine Art. The Migraine Experience From Within. Berkeley, CA: North Atlantic Books (2008).
Google Scholar
18. Carmichael C. Wonderland revisited. London Miscellany. (1996) 28:19–28.
Google Scholar
19. Blom JD. Alice in Wonderland Syndrome., Heidelberg: Springer (2019).
Google Scholar
22. Funatsu N, Hayakawa M, Tokuda N, Toyoda K. Tranzitorii prosopometamorphopsia limitată la ochiul stâng cauzată de ischemie la splenium de corp calos. Intern Med. (2017) 56:2933–5. doi: 10.2169 / internalmedicina.8295-16
PubMed Abstract | CrossRef Textul Complet | Google Scholar
24., Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Caracteristicile variantelor sporadice stabilite și propuse ale bolii Creutzfeldt-Jakob. Arch Neurol. (2009) 66:208-15. doi: 10.1001 / archneurol.2008.533
CrossRef Textul Complet | Google Scholar
26. Heidenhain A. studii clinice și anatomice ale unei boli organice specifice a sistemului nervos central în Praesenium. Z Ges Neurol Psychiat. (1929) 118:49-114. doi: 10.,1007/BF02892896
CrossRef Full Text | Google Scholar
28. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, et al. Revisiting the Heidenhain variant of Creutzfeldt-Jakob disease: evidence for prion type variabiliy influencing clinical course and laboratory findings. J Alzheimers Dis. (2016) 50:465–76. doi: 10.3233/JAD-150668
CrossRef Full Text | Google Scholar
29., Cooper SA, Murray KL, Heath CA, va RG, Cavaler RSG. Simptome vizuale izolate la debut în boala sporadică Creutzfeldt-Jakob: fenotipul clinic al variantei „Heidenhain.”Br J Oftalmol. (2005) 89:1341–2. doi: 10.1136/bjo.2005.074856
PubMed Abstract | CrossRef Textul Complet | Google Scholar