Bakgrunn
Alice in Wonderland syndrom (AIWS) er en sjelden nevrologisk lidelse preget av skjevheter av visuell persepsjon (metamorphopsias), kroppen bildet, og opplevelsen av tid. Som nevnt så tidlig som i 1955 av John Todd, disse symptomene kan være ledsaget av derealisasjon og depersonalisering (1)., Pasienter som lider av AIWS kan ende opp med å konsultere en nevrolog eller psykiater, men i begge spesialiteter det er ikke så godt kjent som den fortjener å være. Dette er i det minste delvis på grunn av det faktum at store grupperinger som Diagnostic and Statistical Manual of Mental Disorders ikke-listen på den som en diagnostisk kategori, mens andre, slik som den Internasjonale statistiske klassifikasjonen av Sykdommer og betaler kun begrenset oppmerksomhet til det. En annen grunn kan være relativt lite antall publiserte saker., En gjennomgang av dokumenterte litteratur, publisert i 2016, indikerte at bare 169 tilfeller av AIWS hadde blitt beskrevet siden syndrom er konseptualisering i 1955, som koker ned til et gjennomsnittlig antall 1.1 tilfeller per år (4). Selv om dette tallet har økt jevnt de siste årene, er det for tiden ligger rundt 180. Av merk, dette er alle saker som har behov for medisinsk tilsyn, dvs., det vi kaller «kliniske tilfeller.»I mellomtiden, stor-skala befolkningen studier tyder på at symptomer på AIWS er opplevd ganske ofte i den generelle befolkningen, med en livstidsprevalens på opp til 30% (5-7)., Siden de sistnevnte tilfeller er vanligvis flyktig og forbigående karakter, og sjelden innebære hjelpe-søkende atferd, de er referert til som «nonclinical tilfeller.»Av de kliniske tilfeller, noen 85% vis involvering av bare en enkelt sensorisk modalitet. Av dem, de fleste er begrenset til et enkelt symptom (f.eks., bare micropsia eller bare macrosomatognosia, etc.) (8). AIWS har mange forskjellige etiologies. I kliniske tilfeller, encefalitt (spesielt på grunn av Epstein-Barr virus-infeksjon) er den vanligste årsaken hos barn; i voksne, det er migrene (4)., Selv om prognose og resultat er i stor grad avhengig av den underliggende sykdommen og dens amenability til behandling, den byrden forårsaket av AIWS kan være betydelig, selv når årsaken er ganske harmløse, som i sammenheng med feber eller sporadisk bruk av cannabis. Dette er spesielt sant når symptomene alvorlig forstyrre ens sensorisk persepsjon, forstyrre ens daglige fungering, og/eller fremkalle katastrofale tanker (for eksempel, at verden har faktisk endret i en uforståelig måte, eller at symptomene kan være tegn på demens eller schizofreni)., Siden ikke-kliniske tilfeller per definisjon er selvbegrensende, og selv i 50% av alle kliniske tilfeller, forsikring synes å være tilstrekkelig, AIWS er noen ganger kan karakteriseres som relativt ufarlig i naturen (9). Imidlertid, siden forskning på dette området er fortsatt i sin spede begynnelse, og AIWS kan godt være sterkt underrapportert, er det for tidlig å trekke noen konklusjoner om sin naturlige historie og «typisk» utfallet. Dessuten, vi vet allerede at AIWS kan være en manifestasjon av alvorlig og til og med dødelig forhold, for eksempel hjerne infarkt eller hjernesvulst (10, 11)., Vi presenterer her et tilfelle av AIWS forårsaket av sporadiske Creutzfeldt-Jakob sykdom (CJD). Med det, vil vi legge til den voksende litteraturen om AIWS, og søker å motvirke noe forhastet konklusjon at dette syndromet kan a priori karakteriseres som relativt «milde» i naturen.
Metoder
pasienten beskriver vi var under behandling i vår egen klinisk praksis. Siden han døde innen 2 måneder etter hans symptomer på AIWS hadde begynt, vi har innhentet skriftlig samtykke for å publisere fra sin familie.,
Resultater–Saken Rapport
I Mars 2018, en 68 år gammel mann som er rapportert til våre nevrologisk poliklinikk med visuelle symptomer. Han hadde en historie av katarakt kirurgi i 2004 og persepsjon døvhet starten i 2006. Et par uker før vi så ham, han hadde vært henvist til øyelege på grunn av dårlig dybde og størrelse persepsjon og en stram følelse rundt hodet som hadde vedvart i 1 måned. Han hadde ingen andre (nevrologiske) symptomer. Ophthalmologic evaluering angitt en åpen vinkel glaukom med intraokulært trykk på 39/27 mmHg ODS – (referanse 12-21 mmHg)., Hans synsskarphet var nesten normal (OD 0·8 dpt, OS 0·9 dpt), selv om det visuelle feltet undersøkelse avdekket flere mild feil (OD) som ikke var i overensstemmelse med hans symptomer. Hans øye press normalisert med bruk av bimatoprost og dorzolamid/timolol øyedråper. Imidlertid, i løpet av de neste ukene, den visuelle symptomer ble mer markant og særegen. Ved henvisning til vår poliklinikk, fortalte han at objekter i bevegelse var å bremse ned og kjøre opp «som i en Charlie Chaplin-filmen» (kjent som langvarig varighet og rask bevegelse fenomen, henholdsvis, dvs.,, varianter av gruppen av tid skjevheter). Noen ganger er han selv ikke klarte å se noen bevegelse i det hele tatt (akinetopsia). I tillegg er han oppfattet farger som usedvanlig lys (hyperchromatopsia) og endre tilfeldig, for eksempel en rød T-skjorte å slå grønne «som messeområdet lys» (chloropsia/dyschromatopsia). Han så også objekter krymping eller svelling opp til en unaturlig størrelse, «som om han var ute i en funhouse speil» (micropsia og macropsia, zoom-visjonen)., I tillegg til disse typer metamorphopsia, utviklet han paraesthesias i hender og føtter, ganglag ustabilitet og konsentrasjon og hukommelse dysfunksjoner. Han hadde også problemer med å spille jazz fløyte og saksofon, som vi betraktet som et illevarslende tegn, gitt at han var en profesjonell jazzmusiker. Hans tale ble mindre flytende fordi han ikke kunne finne de rette ordene, eller unnlatt å string dem sammen til setninger, og hans partner lagt merke til atferdsendring i form av agitasjon og emosjonell labilitet.,
Selv om nevrologisk undersøkelse, rutinemessige blodprøver, og et hode MR (inkludert diffusjon-vektet bilde, DWI) viste ingen avvik, vår pasient ble stadig mer glemsk og desorientert, og ble senere innlagt på våre sykehus. Der så vi en tidligere veltalende mann som nå led av afasi, ataksi, myoclonic rykk, og en hurtig progredierende demens., En annen leder MR viste lave tilsynelatende spredning koeffisient (ADC) verdier og økt DWI-signalering i venstre parieto-occipital cortex, som er karakteristiske for en kortikal båndet tegn (Figur 1), mens elektroencefalogram viste periodisk sharp-wave-komplekser (PSWC) over samme område (Figur 2). MR med tilstedeværelse av 14-3-3 protein og forhøyet tau-protein i spinalvæsken støttes diagnosen sannsynlig sporadiske Creutzfeldt-Jakob sykdom (CJD), særlig Heidenhain variant., Vi utelukket andre mulige årsaker, inkludert auto-immune sykdommer, infeksjoner, malignitet, giftige og metabolske encephalopathies, og andre nevrodegenerativ sykdom (spesielt bakre kortikal atrofi og demens med Lewy legemer). Vi gjorde ikke utføre genetiske tester fordi familien historie var negative for nevrodegenerativ sykdom, CJD i særdeleshet. Dessverre var det ingen terapeutisk valg. I samsvar med hans ønsker, vår pasient ble utskrevet hjem, hvor han døde innen 2 måneder etter hans visuelle symptomer hadde begynt. Vi var i stand til å skaffe obduksjon samtykke.,
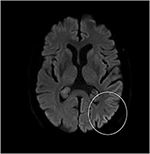
Figur 1. Høyt signal intensitet av venstre parieto-occipital cortex på diffusjon-vektet MR (DWI; b = 1,000).
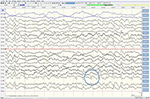
Figur 2. EEG viser periodisk sharp-bølge komplekser over venstre parieto-occipital-regionen, tyder på CJD.,
Diskusjon
Hva er det som gjør denne saken unik, er at vår pasient presentert med et relativt stort antall metamorphopsias (som i den bevarte dokumentasjonen er rapportert i bare 15% av alle kliniske tilfeller av AIWS) og at CJD var den mest sannsynlige årsaken. Siden en diagnose av bestemt CJD kan bare etableres post mortem ved obduksjon, det var uheldig at vi var i stand til å skaffe obduksjon samtykke. Likevel, MR-funn har blitt funnet å være positiv i 83% av tilfellene av sannsynlige CJD (vs. i 98% av tilfellene av bestemt CJD) (12)., Derfor, selv om hodet MR er ikke gullstandarden, en diagnose av CJD var fortsatt veldig sannsynlig i vår pasient, spesielt med tanke på våre andre nevrologiske arbeid-up, som ikke klarte å gi en alternativ diagnose som kan forklare det kliniske bildet, inkludert dens rask nedgang og dramatiske utfall.,
Alice in Wonderland Syndrom
Som beskrevet av John Todd i 1955, AIWS er preget av perseptuelle forstyrrelser som minner om den visuelle forstyrrelser, tid skjevheter, kroppslige endringer, derealisasjon, og depersonalisering oppleves av Alice i løpet av sitt opphold i Wonderland, som beskrevet i Lewis Carroll ‘s klassiske barnebok Alice’ s Adventures in Wonderland (1, 13)., Todd, og før ham, Lipmann (14), foreslo at Carroll–hvis egentlige navn var Charles Lutwidge Dodgson–hadde funnet inspirasjon til disse merkelige fenomener i perseptuelle forstyrrelser han hadde opplevd seg selv i sammenheng med migrene. Selv om denne hypotesen ble etterfulgt av flere historiske tekster med påstand om at migrene var faktisk den mest sannsynlige kilden til Dodgson erfaringer (15, 16), og andre har lagt frem like fristende hypotese som, i hans tilfelle var det heller epilepsi som forårsaket dem (17) eller kanskje rusmisbruk (18)., Selv om vi vil trolig aldri vite sikkert hva som er gjort Dodgson opplever disse symptomene, en nøyaktig rekonstruksjon av sin medisinske historie viser at deres mest sannsynlige kilden var smittsom sykdom, som forfatteren led ved flere anledninger fra barndommen og oppover, bokstavelig talt til den siste dagen i hans liv (19)., Det er svært sannsynlig at Dodgson hadde opplevd disse skjevheter seg selv (heller enn å ha basert sin heltinne er opplevelser på tredje hånd informasjon), med hele 13 ulike symptomer beskrevet gjennom hele boka, lenge før Han hadde kommet opp med konseptet av AIWS.,
for Å forstå hva som gjør AIWS skiller seg ut fra andre perseptuell syndromer og sykdommer, vi må innse at skjevheter forskjellige fra hallusinasjoner og illusjoner i at de er verken nyopprettede percepts av noe som ikke er der (hallusinasjoner), eller faktiske objekter eller scener feilaktig dømt for å være noe annet (illusjon). I stedet, skjevheter er percepts, oppleves av en våken person, som er basert på riktig stimuli fra omverdenen, som en svært spesifikke aspektet er endret på en konsistent måte ., I dag, 55 forskjellige typer av perseptuelle forvrengning er kjent som kan oppstå i sammenheng med AIWS (4). De hyppigst er micropsia (se ting som er mindre enn de er), macropsia (se ting større enn de er), teleopsia (se ting lenger unna enn de er), og dysmorphopsia (se rette linjer som bølgete). Bestemmelsen at slike aspekter er endret på en konsistent måte, betyr at den forvrengning gjelder noe i pasientens visuelle området., Altså, folk som lider av plagiopsia vil vanligvis se alle ting som skjevt, i samme retning, og under samme grad, mens de med prosopometamorphopsia vil vanligvis se en bestemt endring i alle ansikter de oppfatter, som for eksempel tunge øyenbryn eller et øye som er markert bortført til nese eller menneskelige ansikter endring i dragon ansikter (21, 22). Arten av disse skjevheter kan variere over tid, men en gang i dag, alle ting som oppfattes synes som om forvrengt på en lignende måte.

Tabell 1., Definisjoner av hallusinasjoner, illusjoner, og forvrengning .
skillet mellom hallusinasjoner, illusjoner og forvrengning er ikke bare et faglig problem. Tvert imot, deres fenomenologisk forskjellene er tenkt å reflektere forskjeller i underliggende mekanismer. Pathophysiologically, perseptuelle forstyrrelser er knyttet til strukturelle eller funksjonelle lesjoner av diskrete deler av perseptuell-nettverk, for eksempel område V4 i hyperchromatopsia og V5 i akinetopsia (23)., Det er åtte kjente grupper av underliggende etiologi, bestående av smittsomme sykdommer (encefalitt), central nervous system (CNS) lesjoner (hjerneslag, hjernesvulst), perifere nervesystemet (PNS) lesjoner (øyesykdom, midt-øret sykdom), paroksysmal nevrologiske sykdommer (epilepsi, migrene), psykiske lidelser (depresjon, schizofreni), medisiner, illegale stoffer og diverse gruppe som inkluderer hypnagogia og sensorisk deprivasjon (4)., Dermed, såkalte «kliniske tilfeller» av AIWS alltid garanterer diagnostisk arbeid-up, inkludert nevrologisk og psykiatrisk konsultasjon, leder MR og EEG, og, når det er indikert, andre hjelpeorganisasjoner undersøkelser (f.eks., ophthalmologic eksamen, ENT konsultasjon). Selv om evidensbaserte behandlinger som ennå ikke er utviklet, er det god klinisk praksis å sikte terapi på (sannsynlig) underliggende årsak (4, 19).
Creutzfeldt-Jakob Sykdom
Til vår kunnskap, AIWS har ikke blitt rapportert før i sammenheng med CJD. Med 1-1.,5 tilfeller per million innbyggere per år, CJD er en ekstremt sjelden sykdom som er dødelig i løpet av et år i ca 90% av de som berøres (24). Toppen forekomsten er i syvende tiår, mens yngre (20-40 år) eller eldre (>80 år) tilfeller er mye mindre vanlig (25). CJD hører til overførbar spongiform encephalopathies eller prion sykdommer. Prion sykdommer er preget av avleiring av en unormalt misfolded isoformen av de innfødte prion protein, noe som er kodet av prion genet (PRNP) på menneskelig kromosom 20., Mekanismen for å utløse dette conformational endringen er ukjent, men akkumulering av denne unormale prion protein fører til neuronal degenerasjon, astrocytic gliosis, og spongiform endringer, ledsaget av rask kognitiv svikt, og ender i døden.,ximately 85% av tilfellene; (ii) genetisk CJD, som står for 10-15% av tilfellene er assosiert med mutasjoner av prion genet (PRNP); (iii) iatrogenic form av CJD, som står for omtrent 1% av tilfellene, og er oftest forbundet med før behandling med menneskelig hypofyse-avledet hormoner eller menneskelig dura-mater grafts; og (iv) variant CJD (vCJD), som er en roman menneskelige prion sykdom finnes nesten utelukkende i STORBRITANNIA, og har vært knyttet til forbruket av storfekjøtt produkter forurenset med causative agent av bovin spongiform encefalopati (BSE) eller «kugalskap.,»Videre, basert på den første kliniske presentasjon, seks phenotypes har blitt identifisert, kalt kognitiv, visuelle (Heidenhain variant), affektive, klassisk, atactic (Oppenheimer-Brownell variant), og ubestemte (26). Den Heidenhain varianten ble først beskrevet under 1920-tallet, og er oppkalt etter den tyske fysiolog og histologist Rudolf Heidenhain (1834-1897)., Som representerer rundt 20% av alle tilfeller av sporadiske CJD (26-28), det er preget av isolerte visuelle symptomer på sykdom utbruddet, for eksempel visuelle feltet mangler og tap av synsskarphet, som er ansett for å være refleksjoner av tidlig involvering av occipital cortex (29). Siden disse visuelle defekter som kan gå forut for kognitive og motoriske symptomer ved flere uker, og det er ikke uvanlig at CJD er ikke selv er mistenkt i slike tilfeller, og at pasientene er i utgangspunktet henvist til øyelege.,
Konklusjon
Vi konkludere med at muligheten for en Heidenhain variant av CJD, selv om den er meget sjelden, behov for å bli underholdt i differensial diagnose av AIWS, spesielt i nærvær av rask kognitiv svikt. Videre kan vi konkludere med at AIWS kan være relativt ufarlig i de fleste tilfeller, men at «klinisk» saker alltid garanterer et forsvarlig diagnostisk arbeid opp før denne konklusjon kan trekkes.
Begrensninger
Vår kunnskap om de utallige årsaker og manifestasjoner av AIWS er fortsatt i sin spede begynnelse., Syndromet ble først definert i 1955, men bare de siste årene har opplevd en jevn økning i antall publikasjoner på denne uensartede gruppen av fenomener. Siden området er fortsatt svært mye i flux, vår teoretiske refleksjoner på AIWS, selv om state-of-the-art, må derfor betraktes som foreløpige i naturen. For det andre, det faktum at vi presenterer her den første saken rapport om AIWS i sammenheng med CJD betyr ikke nødvendigvis at vår pasient var den første personen til å noensinne lide fra en av disse to sjeldne tilstander., Særlig, Heidenhain variant av CJD kan forventes å presentere oftere med metamorphopsias. Derfor er vår manglende evne til å finne noen før case-beskrivelser av symptomer på AIWS i sammenheng med CJD kan godt være på grunn av underreporting snarere enn til en virkelig sjeldenhet i kombinasjon. Et tredje og siste begrensning er at vi var i stand til å skaffe obduksjon samtykke, som ville ha vært nødvendig for å gjøre en diagnose av bestemt CJD.
Etikk Uttalelse
Skriftlig tillatelse til å publisere ble innhentet fra pasientens familie.,
Bidrag til Feltet Uttalelse
Alice in Wonderland syndrom (AIWS) er en sjelden nevrologisk lidelse preget av skjevheter av visuell persepsjon, kroppen bildet, og opplevelsen av tid. Folk kan se ting som er mindre enn de er, føler at deres kropp endre størrelse, eller opplever noen av de syndrom er en rekke andre symptomer. Siden det er også mange kjente årsaker til AIWS, diagnose krever en grundig nevrologisk arbeid-up. I barn, er den vanligste årsaken er hjernebetennelse, i voksne, det er migrene., I den medisinske litteraturen ikke mer enn 180 enkelte pasienter har blitt beskrevet, 50% av dem tilbake. Imidlertid stor-skala befolkningen studier tyder på at flyktig, forbigående symptomer på AIWS er erfarne med opp til 30% av alle friske individer. Følgelig, AIWS er generelt betraktet som ganske ufarlig i naturen. Vi presenterer her den første saken beskrivelse av en eldre mann som led av AIWS på grunn av Creutzfeldt-Jakob sykdom (CJD), en ekstremt sjelden nevrodegenerativ lidelse., Vår pasient utviklet seg raskt i en tilstand av demens, og døde i løpet av 2 måneder etter hans visuelle symptomer hadde begynt. Vi konkludere med at AIWS er ikke alltid ufarlig, og at CJD, selv om den er meget sjelden, må være på leger sinn når den visuelle symptomer som er typiske for AIWS er ledsaget av rask kognitiv svikt.,
Forfatter Bidrag
TN bidratt til utforming og design av arbeidet, og til innsamling, analyse og tolkning av data for arbeidet, som ble utarbeidet og revidert arbeidet, ga endelig godkjenning for den endelige versjonen til å bli publisert, og ble enige om å være ansvarlig for alle aspekter av arbeidet for å sikre at spørsmål knyttet til nøyaktigheten og integriteten til noen del av arbeidet er behørig undersøkt og løst., BtM, og SvdW bidratt til utforming og design av arbeidet, og til innsamling, analyse og tolkning av data for arbeidet, revidert arbeidet, ga endelig godkjenning for den endelige versjonen til å bli publisert, og ble enige om å være ansvarlig for alle aspekter av arbeidet for å sikre at spørsmål knyttet til nøyaktigheten og integriteten til noen del av arbeidet er behørig undersøkt og løst., JDB bidratt til utforming og design av arbeidet, og til analyse og tolkning av data for arbeidet, som ble utarbeidet og revidert arbeidet, ga endelig godkjenning for den endelige versjonen til å bli publisert, og ble enige om å være ansvarlig for alle aspekter av arbeidet for å sikre at spørsmål knyttet til nøyaktigheten og integriteten til noen del av arbeidet er behørig undersøkt og løst.,
interessekonflikt Uttalelse
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller finansielle forhold som kan oppfattes som en potensiell interessekonflikt.
1. Todd J. syndrom av Alice i Eventyrland. Can Med Assoc J. (1955) 73:701-4.
PubMed Abstrakt | Google Scholar
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5. utg., Washington, DC: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
CrossRef Full Text | Google Scholar
3. World Health Organization. The International Classification of Diseases, 10th Revision. Geneva: World Health Organization (1992).
Google Scholar
5. Abe K, Suzuki T., Utbredelsen av noen symptomer i ungdomsårene og forfall: Sosiale fobier, angst symptomer, episodisk illusjoner og ideen om referanse. Psykopatologi. (1986) 19:200-5. doi: 10.1159/000284448
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar
8. Lanska JR, Lanska DJ. Alice i Eventyrland » – syndrom: somesthetic vs visuelle perseptuelle forstyrrelser. Nevrologi. (2013) 80:1262-4. doi: 10.1212/WNL.,0b013e31828970ae
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar
11. Bender MB, Kanzer MG. Metamorphopsia og andre psychovisual forstyrrelser i en pasient med tumor i hjernen. Arch Neurol Psykiatri. (1941) 45:481-5. doi: 10.1001/archneurpsyc.1941.02280150089005
CrossRef Full Tekst | Google Scholar
13. Carroll L. Alice ‘ s Adventures in Wonderland. London: MacMillan (1865).,
Google Scholar
16. Podoll K, Robinson D. Migraine Art. The Migraine Experience From Within. Berkeley, CA: North Atlantic Books (2008).
Google Scholar
18. Carmichael C. Wonderland revisited. London Miscellany. (1996) 28:19–28.
Google Scholar
19. Blom JD. Alice in Wonderland Syndrome., Heidelberg: Springer (2019).
Google Scholar
22. Funatsu N, Hayakawa M, Tokuda N, Toyoda K. Forbigående prosopometamorphopsia begrenset til venstre øyet forårsaket av iskemi i høyre splenium av corpus callosum. Interne Med. (2017) 56:2933-5. doi: 10.2169/internalmedicine.8295-16
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar
24., Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Egenskaper av etablerte og foreslåtte sporadiske Creutzfeldt-Jakob sykdom varianter. Arch Neurol. (2009) 66:208-15. doi: 10.1001/archneurol.2008.533
CrossRef Full Tekst | Google Scholar
26. Heidenhain A. Kliniske og anatomiske studier av en særegen organisk sykdom i det Sentrale nervesystemet i den Praesenium. Z Location Neurol Psychiat. (1929) 118:49-114. doi: 10.,1007/BF02892896
CrossRef Full Text | Google Scholar
28. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, et al. Revisiting the Heidenhain variant of Creutzfeldt-Jakob disease: evidence for prion type variabiliy influencing clinical course and laboratory findings. J Alzheimers Dis. (2016) 50:465–76. doi: 10.3233/JAD-150668
CrossRef Full Text | Google Scholar
29., Cooper SA, Murray KL Heath CA, Vil RG, Ridder RSG. Isolert visuelle symptomer på utbruddet i sporadiske Creutzfeldt-Jakob sykdom: den kliniske fenotypen av «Heidenhain variant. Br J Ophthalmol. (2005) 89:1341-2. doi: 10.1136/bjo.2005.074856
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar