Baggrund
Alice i Eventyrland syndrom (AIWS) er en sjælden neurologisk lidelse karakteriseret ved forvridninger af visuel perception (metamorphopsias), kroppen billedet, og den oplevelse af tid. Som bemærket allerede i 1955 af John Todd, kan disse symptomer ledsages af derealisering og depersonalisering (1)., Patienter, der lider af AI .s, kan ende med at konsultere en neurolog eller en psykiater, selvom det i begge specialiteter ikke er så kendt som det fortjener at være. Dette skyldes i det mindste delvis, at større klassifikationer som diagnostisk og Statistisk Manual for psykiske lidelser ikke angiver det som en diagnostisk kategori, mens andre, såsom International Classification of Diseases, kun betaler begrænset opmærksomhed på det. En anden grund kan være det relativt lille antal offentliggjorte sager., En gennemgang af den eksisterende litteratur, der blev offentliggjort i 2016, indikerede, at kun 169 tilfælde af AI .s var blevet beskrevet siden syndromets konceptualisering i 1955, hvilket koger ned til et gennemsnitligt antal 1, 1 tilfælde om året (4). Selvom dette antal er steget støt i de sidste par år, ligger det i øjeblikket omkring 180. Det skal bemærkes, at dette alle er tilfælde, der har brug for lægehjælp, dvs.hvad vi kalder “kliniske tilfælde.”I mellemtiden indikerer store befolkningsundersøgelser, at symptomer på AI .s opleves ganske ofte i den generelle befolkning med levetidsprævalensrater på op til 30% (5-7)., Da sidstnævnte tilfælde typisk er flygtige og forbigående, og sjældent medfører hjælpesøgende adfærd, de kaldes “ikke-kliniske tilfælde.”Af de kliniske tilfælde viser nogle 85% involvering af kun en enkelt sensorisk modalitet. Af dem er flertallet begrænset til et enkelt symptom (f.kun mikropsia eller kun makrosomatognosia osv.) (8). AI .s har mange forskellige ætiologier. I kliniske tilfælde er encephalitis (især på grund af Epstein-Barr-virusinfektion) den mest almindelige årsag hos børn; hos voksne er det migræne (4)., Selvom prognose og resultat i vid udstrækning er afhængige af den underliggende lidelse og dens behandlingbarhed, kan byrden forårsaget af AI .s være betydelig, selv når dens årsag er ret ufarlig, som i forbindelse med feber eller sporadisk cannabisbrug. Dette er især tilfældet, når symptomer alvorligt forstyrre ens sansning, forstyrre ens daglige funktion, og/eller fremkalde katastrofale tanker (fx, at verden har faktisk ændret sig i en uforståelig måde, eller at symptomer kan være tegn på demens eller skizofreni)., Da ikke-kliniske tilfælde pr. definition er selvbegrænsende, og selv i 50% af alle kliniske tilfælde synes beroligelse at være tilstrækkelig, er AI .s undertiden karakteriseret som relativt ufarlig i naturen (9). Da forskning på dette område stadig er i sin spædbarn, og AI .s meget vel kan blive underrapporteret, er det for tidligt at drage nogen konklusioner vedrørende dens naturlige historie og “typiske” resultat. Desuden ved vi allerede, at AI .s kan være en manifestation af alvorlige og endda dødelige tilstande, såsom hjerneinfarkt eller hjernesvulst (10, 11)., Vi præsenterer her et tilfælde af AI .s forårsaget af sporadisk Creut .feldt-Jakob sygdom (CJD). Med det tilføjer vi den spirende litteratur om AI .s og søger at imødegå eventuelle for tidlige konklusioner om, at dette syndrom a priori kan karakteriseres som relativt “godartet” i naturen.
metoder
patienten, vi beskriver, var under behandling i vores egen kliniske praksis. Da han døde inden for 2 måneder efter, at hans symptomer på AI .s var begyndt, fik vi skriftligt samtykke til at offentliggøre fra hans familie.,
resultater–Case Report
i Marts 2018 rapporterede en 68-årig mand til vores neurologi ambulant klinik med visuelle symptomer. Han havde en historie med grå stær kirurgi i 2004 og opfattelse døvhed begynder i 2006. Et par uger før vi så ham, var han blevet henvist til øjenlægen på grund af dårlig dybde og størrelsesopfattelse og en stram fornemmelse omkring hovedet, der var vedvarende i 1 måned. Han havde ingen andre (neurologiske) symptomer. Oftalmologisk evaluering indikerede en åbenvinklet glaukom med intraokulært tryk på 39/27 mmHg ODS (reference 12-21 mmHg)., Hans synsskarphed var næsten normal (OD 0·8 dpt, os 0·9 dpt), selvom synsfeltundersøgelsen afslørede flere milde defekter (OD), der ikke var i overensstemmelse med hans symptomer. Hans øjentryk normaliseres ved brug af bimatoprost og dor .olamid/timolol øjendråber. I løbet af de følgende uger blev de visuelle symptomer imidlertid mere udtalt og ejendommelige. Efter henvisning til vores ambulant klinik rapporterede han, at bevægelige genstande bremsede og fremskyndede “som i en Charlie Chaplin-film” (kendt som henholdsvis langvarig varighed og hurtigbevægelsesfænomen, dvs.,, varianter af gruppen af tidsforvrængninger). Nogle gange kunne han endda ikke se nogen bevægelse overhovedet (akinetopsia). Hertil kommer, at han opfattede farver, som undtagelsesvis lyse (hyperchromatopsia) og ændring tilfældigt, såsom en rød T-shirt dreje grønne “som tivoli-lys” (chloropsia/dyschromatopsia). Han så også genstande krympe eller hæve op til en unaturlig størrelse, “som om han kiggede ind i et funhouse spejl” (micropsia og macropsia, visionoom vision)., Ud over disse typer af metamorfopsi udviklede han paræstesier i hænder og fødder, gangstabilitet og koncentration og hukommelsesdysfunktioner. Han havde også svært ved at spille Ja.flfløjte og Sa .ofon, som vi betragtede som et ildevarslende tegn, da han var en professionel Ja. .musiker. Hans tale blev mindre flydende, fordi han ikke kunne finde de rigtige ord eller undlod at strenge dem sammen for at lave sætninger, og hans partner bemærkede adfærdsændringer i form af agitation og følelsesmæssig labilitet.,
Selv om neurologisk undersøgelse, rutinemæssige blodprøver, og et hoved, MR (herunder diffusion-vægtet imaging, DWI) viste ingen abnormiteter, vores patient blev mere og mere glemsom og desorienteret, og blev efterfølgende indlagt på vores hospital. Der så vi en tidligere veltalende Mand, der nu led af afasi, ataksi, myokloniske rykker og en hurtigt progressiv demens., En anden chef MR-scanning viste en lav tilsyneladende diffusion coefficient (ADC) værdier og øget DWI-signalering i venstre parieto-occipital cortex, som er karakteristisk for en kortikale bånd tegn (Figur 1), mens elektroencefalogram viste periodiske skarpe-bølge komplekser (PSWC) over det samme område (Figur 2). MR med tilstedeværelse af 14-3-3 protein og forhøjet tau protein i cerebrospinalvæsken, der understøttes diagnosen sandsynlig sporadisk Creutzfeldt-Jakobs sygdom (CJD), især Heidenhain-variant., Vi udelukket andre mulige årsager, herunder auto-immune sygdomme, infektioner, maligne sygdomme, toksiske og metaboliske encephalopatier, og andre neurodegenerative sygdomme (især posterior kortikal atrofi og demens med Lewy legemer). Vi udførte ikke genetisk test, fordi familiehistorien var negativ for neurodegenerativ sygdom, især CJD. Desværre var der ingen terapeutiske muligheder. I overensstemmelse med hans ønsker blev vores patient udskrevet hjem, hvor han døde inden for 2 måneder efter, at hans visuelle symptomer var begyndt. Vi kunne ikke få tilladelse til obduktionen.,
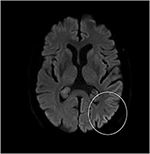
Figur 1. Høj signalintensitet af venstre parieto-occipital Corte.på diffusionsvægtet Mr (D .i; B = 1.000).
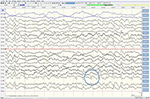
Figur 2. EEG viser periodiske skarpbølgekomplekser over den venstre parieto-occipitale region, der tyder på CJD.,
Diskussion
Hvad gør dette tilfælde unikt, er, at vores patient præsenteret med et relativt stort antal metamorphopsias (som i den eksisterende litteratur, er rapporteret i kun 15% af alle kliniske tilfælde af AIWS), og at CJD var den mest sandsynlige årsag. Da en diagnose af bestemt CJD kun kan etableres post mortem ved obduktion, det var uheldigt, at vi ikke var i stand til at opnå obduktions samtykke. Alligevel har MR-fund vist sig at være positive i 83% af tilfældene med sandsynlig CJD (vs. i 98% af tilfældene med bestemt CJD) (12)., Derfor, selvom hoved MR ikke er guldstandarden, var en diagnose af CJD stadig meget sandsynlig hos vores patient, især i betragtning af vores yderligere neurologiske oparbejdning, som ikke gav en alternativ diagnose, der kunne forklare det kliniske billede, herunder dets hurtige tilbagegang og dramatiske resultat.,
Alice i Eventyrland-Syndromet
Som beskrevet af John Todd i 1955, AIWS er præget af perceptuelle forvridninger, der minder om den visuelle skævheder, tid forvridninger, kropslige ændringer, derealisation og depersonalisation oplevet af Alice, under hendes ophold i Eventyrland, som beskrevet i Lewis carrolls klassiske børnebog Alice ‘ s Adventures in Wonderland (1, 13)., Todd, og før ham, Lipmann (14), foreslog, at Carroll–hvis rigtige navn var Charles Lutwidge Dodgson–havde fundet inspiration til disse ejendommelige fænomener i perceptuelle forvridninger, han havde oplevet sig selv i forbindelse med migræne. Selv om denne hypotese blev fulgt af adskillige historiske afhandlinger med påstand om, at migræne var faktisk den mest sandsynlige kilde til Dodgson oplevelser (15, 16), og andre har fremsat den lige så vage hypotese, at det i hans tilfælde var det snarere epilepsi, der er forårsaget dem (17) eller måske stofmisbrug (18)., Selvom vi sandsynligvis aldrig vil vide med sikkerhed, hvad der fik Dodgson til at opleve disse symptomer, indikerer en omhyggelig rekonstruktion af hans medicinske historie, at deres mest sandsynlige kilde var infektionssygdom, hvorfra forfatteren led ved adskillige lejligheder fra barndommen og fremefter, bogstaveligt talt indtil den sidste dag i hans liv (19)., Det er meget sandsynligt, at Dodgson havde oplevet disse forvridninger sig selv (snarere end at have baseret sin heltinde ‘ s oplevelser på tredje hånd oplysninger), med en kæmpestor 13 forskellige symptomer, der er beskrevet i hele bogen, længe før Todd var kommet op med begrebet AIWS.,
for At forstå, hvad der gør AIWS skiller sig ud fra andre perceptuelle syndromer og lidelser, vi er nødt til at indse, at forvridninger adskiller sig fra hallucinationer og illusioner i, at de hverken er nyligt dannet percepts af noget, der ikke er der (hallucinationer), eller faktiske objekter og scener, fejlagtigt dømt til at være noget andet (illusion). I stedet er forvrængninger percepter, der opleves af et vågent individ, som er baseret på passende stimuli fra omverdenen, hvoraf et meget specifikt aspekt ændres på en konsistent måde ., I dag er der kendt omkring 55 forskellige typer perceptuel forvrængning, der kan forekomme i forbindelse med AI .s (4). De hyppigst rapporterede er mikropsia (ser ting mindre end de er), makropsia (ser ting større end de er), teleopsia (ser ting længere væk end de er) og dysmorphopsia (ser lige linjer som bølget). Bestemmelsen om, at sådanne aspekter ændres på en konsistent måde, betyder, at forvrængningen gælder for alt inden for patientens visuelle rækkevidde., Således mennesker, der lider af plagiopsia vil typisk se alle ting, som skrå, i samme retning, og under samme grad, mens dem med prosopometamorphopsia vil typisk se en bestemt ændring i alle de ansigter, de opfatter, såsom tunge øjenbryn eller et øje, der er markant bortført til næsen eller ansigter på at ændre i dragon ansigter (21, 22). Arten af disse forvrængninger kan variere over tid, men når de først er til stede, ser alle ting, der opfattes, ud som forvrænget på en lignende måde.

Tabel 1., Definitioner af hallucination, illusion og forvrængning .
sondringen mellem hallucination, illusion og forvrængning er ikke blot et akademisk spørgsmål. Tværtimod menes deres fænomenologiske forskelle at afspejle forskelle i underliggende mekanismer. Patofysiologisk, perceptuelle-fordrejninger tilskrives strukturelle eller funktionelle læsioner i diskrete dele af den perceptuelle netværk, såsom område V4 i hyperchromatopsia og V5 i akinetopsia (23)., Der er otte kendte grupper af underliggende ætiologi, bestående af smitsomme sygdomme (encephalitis), centralnervesystem (CNS) læsioner (hjerneblødning, hjernesvulst), perifere nervesystem (PNS), læsioner (øjensygdom, midt-øre-sygdomme), paroxysmal neurologiske sygdomme (epilepsi, migræne), psykiske lidelser (depression, skizofreni), medicin, illegale stoffer, og en diverse-gruppen, som omfatter hypnagogia og sensorisk deprivation (4)., Således, såkaldt “kliniske tilfælde” af AIWS altid garanterer diagnostiske arbejde-up, herunder neurologiske og psykiatriske konsultation, hoved MR-scanning og EEG, og, når det er angivet, andre supplerende undersøgelser (fx, oftalmologisk undersøgelse, ENT høring). Selvom evidensbaserede behandlinger endnu ikke er udviklet, er det god klinisk praksis at sigte terapi mod den (sandsynlige) underliggende årsag (4, 19).
Creut .feldt-Jakob sygdom
efter vores viden er AI .s ikke blevet rapporteret før i forbindelse med CJD. Med 1-1.,5 tilfælde pr. million befolkning om året, CJD er en ekstremt sjælden sygdom, der er dødelig inden for et år hos omkring 90% af de berørte (24). Topincidensen er i det syvende årti, mens yngre (20-40 år) eller ældre (>80 år) tilfælde er meget mindre almindelige (25). CJD hører til de transmissible spongiforme encephalopatier eller prionsygdomme. Prionsygdomme er kendetegnet ved aflejring af en unormalt forkert foldet isoform af det native prionprotein, som kodes af priongenet (PRNP) på humant kromosom 20., Mekanismen til at udløse denne konformationsændring er ukendt, men akkumuleringen af dette unormale prionprotein fører til neuronal degeneration, astrocytisk gliose og spongiforme ændringer ledsaget af hurtig kognitiv tilbagegang og slutter i døden.,ximately 85% af de tilfælde; (ii) genetiske CJD, som tegner sig for 10-15% af tilfældene og er forbundet med mutationer af prion-gen (PRNP); (iii) de iatrogene form af CJD, som tegner sig for omkring 1% af alle tilfælde og er oftest forbundet med forudgående behandling med human hypofyse fremstillet på basis af hormoner eller menneskelige dura-mater grafts; og (iv) variant af CJD (vCJD), som er en roman menneskelige prionsygdom findes næsten udelukkende i det forenede KONGERIGE, og har været knyttet til forbrug af oksekødsprodukter forurenet med det agens af bovin spongiform encephalopati (BSE) eller “kogalskab.,”Baseret på den indledende kliniske præsentation er der desuden identificeret seks fænotyper, kaldet kognitiv, visuel (Heidenhain-variant), affektiv, klassisk, ataktisk (Oppenheimer-bro .nell-variant) og ubestemt (26). Heidenhain-varianten blev først beskrevet i 1920 ‘ erne og opkaldt efter den tyske fysiolog og histolog Rudolf Heidenhain (1834-1897)., 20% af alle tilfælde af sporadisk CJD (26-28), det er kendetegnet ved isolerede visuelle symptomer ved sygdomsudbrud, såsom synsfeltdefekter og tab af synsskarphed, der betragtes som refleksioner af tidlig involvering af occipital Corte. (29). Da disse visuelle defekter kan gå forud for kognitive og motoriske symptomer med flere uger, er det ikke ualmindeligt, at CJD ikke engang mistænkes i sådanne tilfælde, og at patienter oprindeligt henvises til en øjenlæge.,
konklusion
vi konkluderer, at muligheden for en Heidenhain-variant af CJD, selvom den er ekstremt sjælden, skal underholdes i den differentielle diagnose af AI .s, især i nærvær af hurtig kognitiv tilbagegang. Desuden konkluderer vi, at AI .s kan være relativt harmløse i de fleste tilfælde, men at “kliniske” tilfælde altid berettiger en ordentlig diagnostisk oparbejdning, før denne konklusion kan drages.
begrænsninger
vores viden om de mange årsager og manifestationer af AI .s er stadig i sin spædbarn., Syndromet blev først konceptualiseret i 1955, men kun de sidste par år har været vidne til en jævn stigning i antallet af publikationer om denne forskellige gruppe af fænomener. Da området stadig er meget i Flu., skal vores teoretiske refleksioner om AI .s, selvom de er avancerede, betragtes som foreløbige. For det andet betyder det faktum, at vi her præsenterer den første sagsrapport om AI .s i forbindelse med CJD, ikke nødvendigvis, at vores patient var den første person, der nogensinde lider af disse to sjældne tilstande., Især kan Heidenhain-varianten af CJD forventes at præsentere hyppigere med metamorphopsier. Derfor, vores manglende evne til at finde nogen forudgående sagsbeskrivelser af symptomer på AI .s i forbindelse med CJD kan meget vel skyldes underrapportering snarere end en faktisk sjældenhed af kombinationen. En tredje og sidste begrænsning er, at vi ikke var i stand til at opnå obduktions samtykke, hvilket ville have været nødvendigt for at stille en diagnose af bestemt CJD.
etisk Erklæring
skriftligt samtykke til offentliggørelse blev opnået fra patientens familie.,
Bidrag til Feltopgørelsen
Alice in Wonderonderland syndrom (AI .s) er en sjælden neurologisk lidelse præget af forvrængninger af visuel opfattelse, kropsbilledet og oplevelsen af tid. Folk kan se tingene mindre end de er, føler deres krop ændre sig i størrelse eller opleve nogen af syndromets mange andre symptomer. Da der også er mange kendte årsager til AI .s, kræver diagnosen en grundig neurologisk oparbejdning. Hos børn er den mest almindelige årsag hjernebetændelse; hos voksne er det migræne., I den medicinske litteratur er der ikke beskrevet mere end 180 individuelle patienter, hvoraf 50% blev genoprettet. Imidlertid viser store befolkningsundersøgelser, at flygtige, forbigående symptomer på AI .s opleves af op til 30% af alle sunde individer. Derfor betragtes AI .s generelt som temmelig ufarlig i naturen. Vi præsenterer her den første case beskrivelse af en ældre mand, der led af AIWS på grund af Creutzfeldt-Jakobs sygdom (CJD), en yderst sjælden neurodegenerative lidelse., Vores patient udviklede hurtigt en tilstand af demens og døde inden for 2 måneder efter, at hans visuelle symptomer var begyndt. Vi konkluderer, at AI .s ikke altid er ufarlig, og at CJD, selvom det er ekstremt sjældent, skal være på lægernes sind, når de visuelle symptomer, der er typiske for AI .s, ledsages af hurtig kognitiv tilbagegang.,
Forfatter Bidrag
TN bidraget til udformning og design af de arbejder, og til indsamling, analyse og tolkning af data for det arbejde, der er udarbejdet og revideret arbejde, gav endelig godkendelse, til den endelige version, der skal offentliggøres, og blev enige om at være ansvarlige for alle aspekter af arbejdet med at sikre, at spørgsmål vedrørende nøjagtigheden eller integriteten af nogen del af det arbejde, der er tilstrækkeligt undersøgt og løst., BtM, og SvdW bidraget til udformning og design af de arbejder, og til indsamling, analyse og tolkning af data for det arbejde, revideret arbejde, gav endelig godkendelse, til den endelige version, der skal offentliggøres, og blev enige om at være ansvarlige for alle aspekter af arbejdet med at sikre, at spørgsmål vedrørende nøjagtigheden eller integriteten af nogen del af det arbejde, der er tilstrækkeligt undersøgt og løst., JDB bidraget til udformning og design af de arbejder, og til den analyse og fortolkning af data for det arbejde, der er udarbejdet og revideret arbejde, gav endelig godkendelse, til den endelige version, der skal offentliggøres, og blev enige om at være ansvarlige for alle aspekter af arbejdet med at sikre, at spørgsmål vedrørende nøjagtigheden eller integriteten af nogen del af det arbejde, der er tilstrækkeligt undersøgt og løst.,
Erklæring om interessekonflikt
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
1. Todd J. syndromet af Alice i Eventyrland. Kan Assith Assoc J. (1955) 73:701-4.
PubMed Abstract | Google Scholar
2. American Psychiatric Association. Diagnostisk og Statistisk Manual for psykiske lidelser. 5. udgave., Washington, DC: American Psychiatric Association (2013). doi: 10.1176/appi.books.9780890425596
CrossRef Full Text | Google Scholar
3. World Health Organization. The International Classification of Diseases, 10th Revision. Geneva: World Health Organization (1992).
Google Scholar
5. Abe K, Suzuki T., Udbredelse af nogle symptomer i ungdomsårene og modenhed: sociale fobier, angstsymptomer, episodiske illusioner og ID.om reference. Psykopatologi. (1986) 19:200–5. doi: 10.1159/000284448
PubMed Abstract | CrossRef Fuld Tekst | Google Scholar
8. Lanska JR, Lanska DJ. Alice i Eventyrland syndrom: nogleæstetisk vs visuel perceptuel forstyrrelse. Neurologisk. (2013) 80:1262–4. doi: 10.1212/WNL.,0b013e31828970ae
PubMed Abstract | CrossRef Fuld Tekst | Google Scholar
11. Bender MB, kan MGER MG. Metamorfopsi og andre psyko-synsforstyrrelser hos en patient med hjernetumor. Arch Neurol Psykiatri. (1941) 45:481–5. doi: 10.1001/archneurpsyc.1941.02280150089005
CrossRef Fuld Tekst | Google Scholar
13. Carroll L. Alice eventyr i Eventyrland. London: MacMillan (1865).,
Google Scholar
16. Podoll K, Robinson D. Migraine Art. The Migraine Experience From Within. Berkeley, CA: North Atlantic Books (2008).
Google Scholar
18. Carmichael C. Wonderland revisited. London Miscellany. (1996) 28:19–28.
Google Scholar
19. Blom JD. Alice in Wonderland Syndrome., Heidelberg: Springer (2019).
Google Scholar
22. Funatsu N, Hayakawa M, Tokuda N, Toyoda K. Forbigående prosopometamorphopsia begrænset til det venstre øje, der er forårsaget af iskæmi i den rigtige splenium af corpus callosum. Praktikant Med. (2017) 56:2933–5. doi: 10.2169/internalmedicin.8295-16
PubMed Abstract | CrossRef Fuld Tekst | Google Scholar
24., Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Karakteristik af etablerede og foreslåede sporadiske Creut .feldt-Jakob sygdomsvarianter. Arch Neurol. (2009) 66:208-15. doi: 10.1001/archneurol.2008.533
CrossRef Fuld Tekst | Google Scholar
26. Heidenhain A. kliniske og anatomiske undersøgelser af en ejendommelig organisk sygdom i centralnervesystemet i Praeseniet. Z Ges Neurol Psychiat. (1929) 118:49-114. doi: 10.,1007/BF02892896
CrossRef Full Text | Google Scholar
28. Baiardi S, Capellari S, Ladogana A, Strumia S, Santangelo M, Pocchiari M, et al. Revisiting the Heidenhain variant of Creutzfeldt-Jakob disease: evidence for prion type variabiliy influencing clinical course and laboratory findings. J Alzheimers Dis. (2016) 50:465–76. doi: 10.3233/JAD-150668
CrossRef Full Text | Google Scholar
29., Cooper SA, Murray KL, Heath CA, vil RG, ridder RSG. Isolerede visuelle symptomer ved indtræden i sporadisk Creut .feldt-Jakob-sygdom: den kliniske fænotype af “Heidenhain-varianten.”Br J Ophthalmol. (2005) 89:1341–2. doi: 10.1136 / bjo.2005.074856
PubMed Abstract | CrossRef Fuld Tekst | Google Scholar